Alinhamento
Apesar de muitos sistematas darem pouca importância ao alinhamento e o tratarem apenas como parte do processo para a obtenção de uma árvore filogenética, esta etapa é extremamente importante para a análise filogenética como um todo, pois é a etapa em que definimos a hipótese de homologia primária entre as nossas sequências. Em outras palavras, nesta etapa assumimos que as bases que estão alinhadas em uma mesma coluna divergiram de um mesmo estado ancestral. Sendo esta uma etapa tão importante, existem vários softwares e algoritmos que são comumente utilizados para alinhar sequências de DNA (e também de aminoácidos, mas não vamos abordar esta parte no curso), como por exemplo ClustalW (Larkin et al., 2007), T-Coffee (Notredame et al., 2000), MUSCLE (Edgard, 2004) e MAFFT (Katoh & Standley, 2013). No nosso curso abordaremos somente os dois últimos.
MUSCLE (implementado no Mega7)
Voltando ao seu trabalho com a filogenia de Hylodidae do item anterior. Agora que você adquiriu as sequências de 16S rRNA e COI no GenBank você precisa alinhá-las. Para te poupar o trabalho de filtrar o mundo de sequências baixadas, preparamos versões com um exemplar por espécie apenas tanto de 16S quanto de COI. Também adicionamos uma sequência de Limnomedusa macroglossa para servir como raiz da sua árvore nas futuras análises.
Vamos primeiro abrir as sequências de COI no software MEGA. Está tudo desalinhado. Selecione todas as sequências (com ctrl + a) e então vá em:
Alignment > Align By Muscle
Uma tela irá se abrir, onde você poderá mudar alguns parâmetros no algoritmo MUSCLE. Vamos utilizar o default, clique no botão Compute. Agora aguarde um pouquinho e pronto! Suas sequências estão alinhadas.
O COI é um gene codificante, então algo interessante a se fazer é ver se as sequências estão corretas, começando no codon certo, ou se os possíveis gaps são múltilos de três, por exemplo. Isso influenciará em etapas posteriores, então é interessante que sempre comecemos no codon 1. Isso também serve para checar se não existem stop codons no meio das suas sequências, o que poderia indicar que você sequenciou um pseudogene ou o gene errado. Após o alinhamento pronto, clique em Translated Protein Sequences, e então quando a caixa se abrir com a pergunta The Current Genetic Code is: Standard. Is this correct? clique em No, como na figura abaixo:
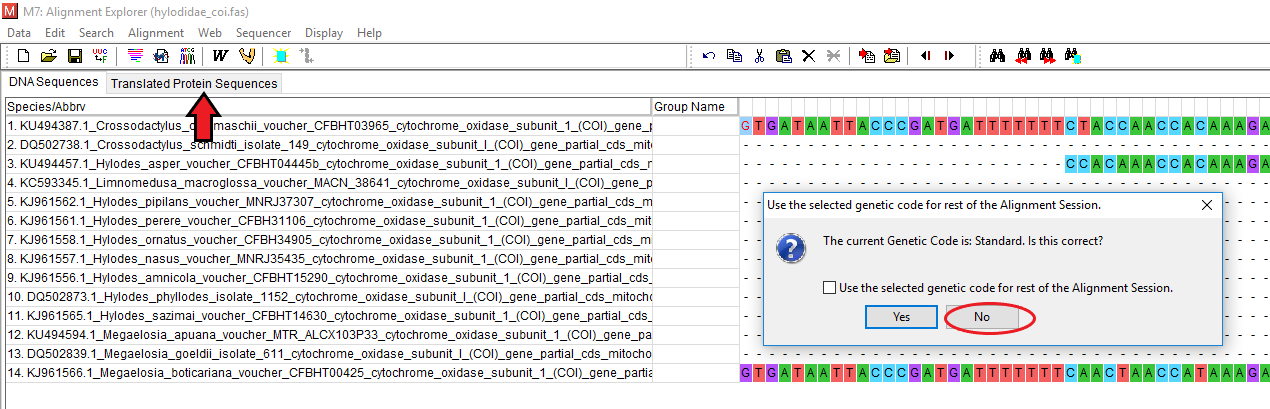
Então outra caixa se abrirá e, já que se trata do fragmento COI de anfíbios marque a opção Vertebrate mitochondrial:
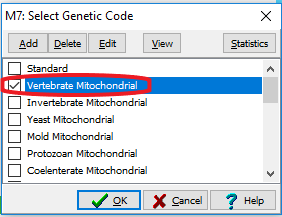
Agora é só ver se não há stop codons (indicado pelo símbolo *) em nenhuma das posições. Se houver, provavelmente seu arquivo não começa em uma posição 1 do códon ou o gene que você sequenciou não é o que você queria.
Repita o processo com as sequências de 16S mas, visto que este não é um gene codificante, não faz sentido traduzir as sequências. Observe seu alinhamento: qual a principal diferença entre este alinhamento e o do COI? Qual dos dois alinhamentos demorou mais para ser concluído? Por que?
MAFFT (usando localmente)
O MAFFT é um programa um pouco mais completo que o MUSCLE, por ter vários algoritmos diferentes para alinhar suas sequências. Alguns deles são mais rápidos e menos precisos, ideais para um grande número de sequências ou para fragmentos codificantes, sem estrutura secundária, e outros são mais precisos e mais lentos, e são ideais para fragmentos com estrutura secundária, por exemplo. Por serem mais lentos, estes últimos não podem ser utilizados em grandes conjuntos de sequências.
O que devemos fazer primeiramente é copiar os dois arquivos com as sequências que baixamos acima no diretório em que o programa mafft está instalado. Feito isto, abra o arquivo mafft.bat no diretório em que você instalou o programa. Uma janela do prompt de comando se abrirá e você deverá digitar o nome do arquivo a ser alinhado. Vamos começar pelas sequências de COI:
MAFFT v7.409 (2018/Aug/22)
MBE 30:772-780 (2013), NAR 30:3059-3066 (2002)
https://mafft.cbrc.jp/alignment/software/
---------------------------------------------------------------------
Input file? (FASTA format; Folder="C:\Users\Pedro Taucce\Documents\mafft-win")
@ hylodidae_coi.fas
Se correu tudo bem, o programa deve mostrar a seguinte mensagem:
OK. infile = hylodidae_coi.fas
Agora devemos escrever o nome pretendido do arquivo em que teremos as sequências alinhadas. Não escreva o mesmo nome ou o programa não vai funcionar! Assim:
Output file?
@ hylodidae_coi_alinhado.fas
Nossa próxima escolha será em qual formato o nosso arquivo de saída estará. Vamos continuar no formato .fasta, e escolhendo a opção 3 colocaremos as sequências parecidas mais próximas umas das outras. Isto pode ser interessante para vermos se há alguma anomalia nas nossas sequências ou no nosso alinhamento.
Output format?
1. Clustal format / Sorted
2. Clustal format / Input order
3. Fasta format / Sorted
4. Fasta format / Input order
5. Phylip format / Sorted
6. Phylip format / Input order
@ 3
Depois temos que escolher qual algoritmo de alinhamento usaremos. Escolhendo a opção 1 o programa escolhe um dos algoritmos automaticamente. Como conhecemos o programa, escolheremos a opção dois FFT-NS-2, que parte de uma matriz de distâncias confiável mas não é tão lenta quanto outros algoritmos presentes no MAFFT.
Strategy?
1. --auto
2. FFT-NS-1 (fast)
3. FFT-NS-2 (default)
4. G-INS-i (accurate)
5. L-INS-i (accurate)
6. E-INS-i (accurate)
@ 3
Por último, podemos adicionar outros parâmetros. Adicionaremos apenas um, para que as bases das sequências sejam mantidas em letra maiúscula (--preservecase). Depois é só apertar a tecla enter. Quando o programa terminar, aperte a letra q e então a tecla enter.
Additional arguments? (--ep # --op # --kappa # etc)
@ --preservecase
command=
"C:/Users/Pedro Taucce/Documents/mafft-win/usr/bin/mafft" --preservecase --retree 2 --reorder "hylodidae_coi.fas" > "hylodidae_coi_alinhado.fas"
OK?
@ [Y]
Agora vamos fazer a mesma coisa com o fragmento 16S. com apenas algumas diferenças. Na hora de escolhermos o algoritmo, escolheremos o número 6, E-INS-i, que lida bem com fragmentos como este. Também adicionaremos o parâmetro --maxiterate 1000, pois este algoritmo faz iterações e vai tentando refinar o alinhamento até conseguir um alinhamento satisfatório. Com este parâmetro aumentaremos o número de iterações possíveis para 1000. Teremos as seguintes diferenças co relação ao alinhamento anterior:
1. --auto
2. FFT-NS-1 (fast)
3. FFT-NS-2 (default)
4. G-INS-i (accurate)
5. L-INS-i (accurate)
6. E-INS-i (accurate)
@ 3
Additional arguments? (--ep # --op # --kappa # etc)
@ --preservecase --maxiterate 1000
Caso queira saber mais sobre os algoritmos do MAFFT é só clicar aqui.
MAFFT (no servidor)
Algumas vezes possuimos um número muito grande de sequências muito longas, e fazer o alinhamento no nosso computador pode demorar horas e até mesmo dias. Para momentos como este (ou até para ser utilizado em computadores que não possuem MAFFT instalado) existe um servidor online, mantido pelo próprio grupo que desenvolveu o software. Vamos alinhar o fragmento do 16S nesse servidor.
Primeiramente entre no site do servidor. Você verá uma caixinha para colar as sequências (como fizemos no BLAST) e também um botão escolher arquivo. Clique nele e suba o arquivo hylodidae_16S.fas para o servidor, e depois marque as opções como na figura abaixo:
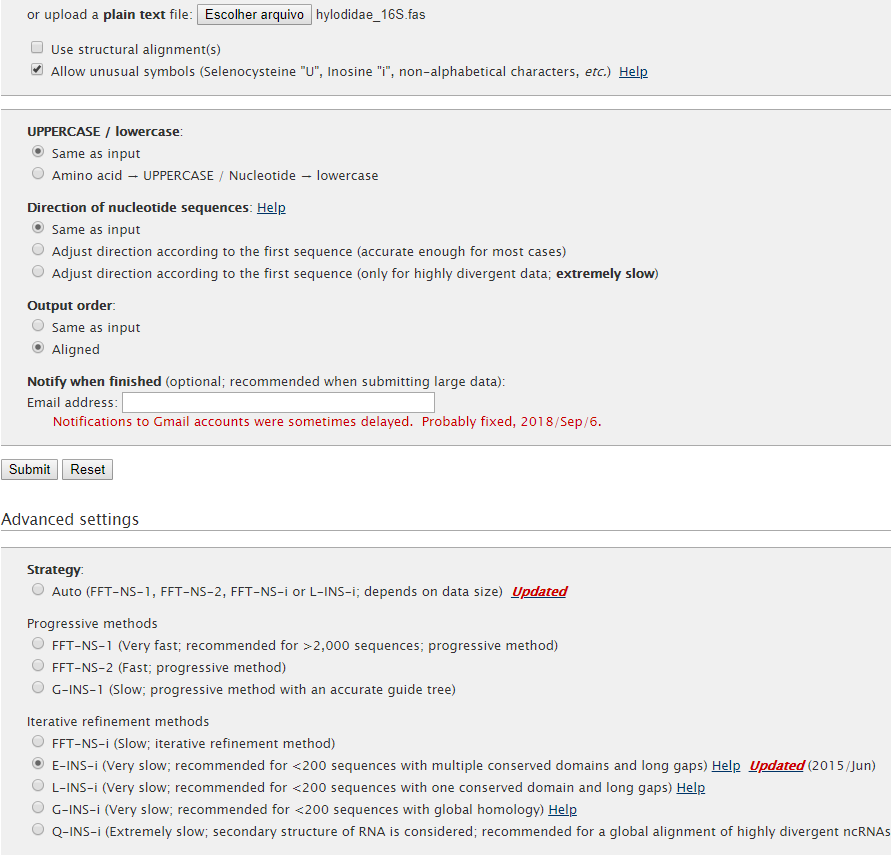
Não é necessário mexer nos outros parâmetros, deixe-os no default. Com isto faremos um alinhamento parecido com o que fizemos no item anterior. Agora clique no botão submit.
Agora o navegador irá para outra página, com os resultados. Para baixar o arquivo resultante como .fasta é só clicar com o botão direito onde está escrito Fasta format e depois em Salvar link como. Salve o arquivo resultante com o nome que quiser, mas use a extensão .fas.
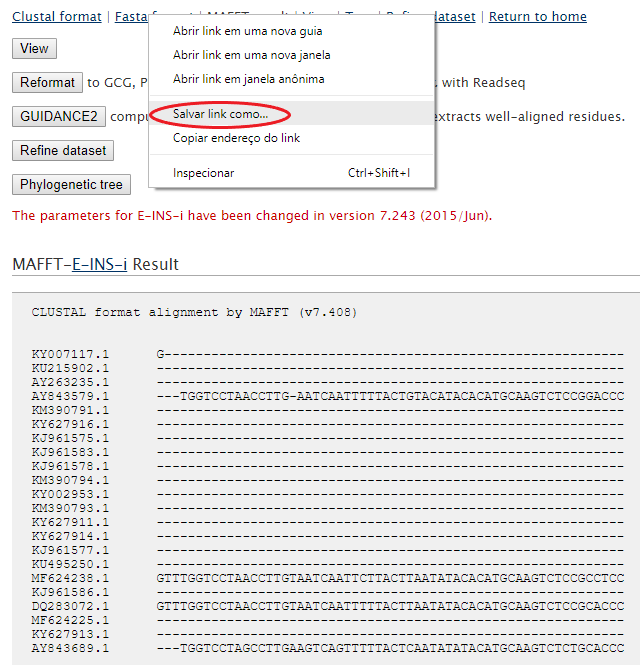
Agora com os alinhamentos terminados podemos avançar e ir concatenar a matriz!
Referências
Edgar RC. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792-1797.
Katoh K, Standley DM. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772-780.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947-2948.
Notredame C, Higgins DG, Heringa J. (2000). T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. Journal of Molecular Biology, 302,205-217.