Sequence Matrix
Vamos abordar aqui apenas como juntar diferentes fragmentos em um mesmo terminal para concatenar seus dados. Este programa reconhece diversos formatos de arquivos de sequências: .fas, .tnt, .nex, etc… A primeira coisa é ir no Menu superior:
Import > Add sequences
E então adicionar o primeiro arquivo desejado. Dependendo do formato dos nomes dos terminais, se por exemplo vierem acrescidos de números de tombo (ou número de acesso no GenBank, etc.), uma caixa de diálogo abrirá, perguntando se você deseja que os terminais saiam com o número, ou apenas o nome das espécies.
Se você tem mais de um terminal para uma mesma espécie, é desejável que mantenha a sua nomenclatura original (i.e., com o numero da amostra) para saber onde esta amostra estará em sua futura árvore. Então clique em “Use sequence names”. Em seguida, ou se seus terminais originalmente continham apenas o nome da espécie no nome, a seguinte caixa de diálogo abrirá:
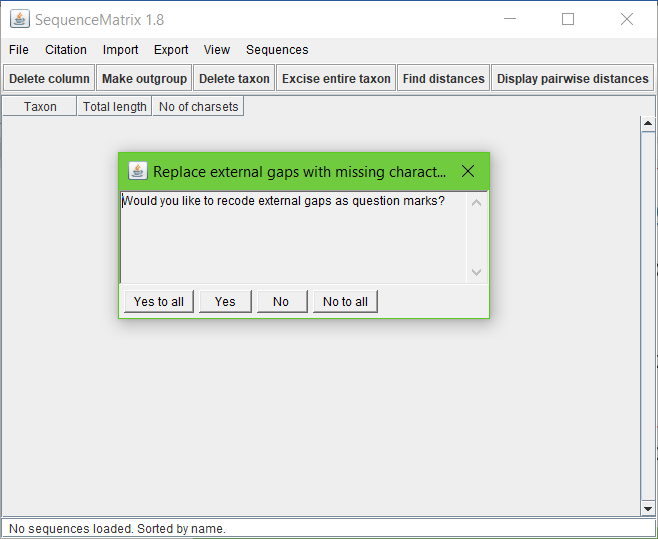
Clique em “Yes to all”. A não ser que não deseje isto para sua futura análise.
Repita o procedimento de importação até obter todos os fragmentos de DNA que deseja incluir em sua matriz. P.S.: é importante, e também mais prático, que você tenha um arquivo por tipo fragmento. Por exemplo, um arquivo fasta com todas as sequências do mitocondrial 12S, um arquivo fasta com todas as sequências do nuclear RAG1, e etc. Ao terminar as importações, você deverá ter algo semelhante à seguinte matriz:
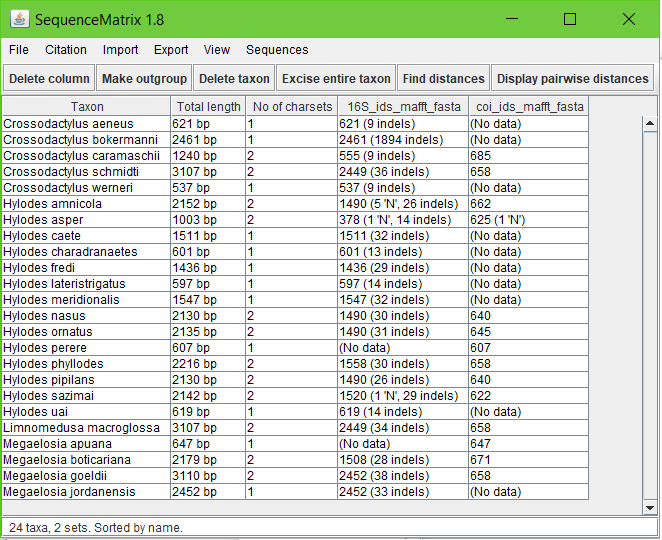
Nela, a primeira coluna (táxon; da esquerda para a direita) corresponde aos seus terminais (o Sequence Matrix sempre organiza em ordem alfabética). A segunda (total length), o total de nucleotídeos por terminal, a terceira (No of charsets) indica o total de fragmentos que cada terminal possui de fato, dentre os X que você está adicionando). As subsequentes colunas são os distintos fragmentos que você importou.
É muito importante, que em cada arquivo os nomes dos terminais estejam idênticos, caso o contrário, o programa reconhecerá como dois ou mais táxons distintos. Por exemplo. Imagine uma matriz com 12S e RAG1. No arquivo de 12S, o terminal está redigido como:
Dugong_dugon_MCZ123
Enquanto que no arquivo RAG1, o terminal está como:
Dugongo_dugon_MCZ123
Ao importar, o programa reconhecerá como dois terminais, tendo 1 charset cada, apenas, graças ao “o” extra no segundo arquivo. Sendo que, você desejaria que os dois fragmentos correspondessem ao mesmo terminal. Para solucionar isso, clique duas vezes sobre o nome redigido errado, e uma barra de texto surgirá. Ao corrigir, se você redigiu corretamente, o programa automaticamente juntará as duas sequências em um único terminal.
Uma vez que tenha importado todas as sequências e corrigido os nomes errados, agora basta exportar. Menu superior Export. Selecione o formato de arquivo desejado, e uma caixa se abrirá perguntando onde quer salvá-lo.
Experimente exportar os três seguintes diferentes formatos de arquivos:
hylodidae.tnt (opção “Export sequences as TNT”)
hylodidae.nex (opção “Export sequences as NEXUS [“naked” e.g. for GARLI])
hylodidae.phy (opção “Export sequences for RAxML analyses on CIPRES”)
*é importante ao digitar o nome do arquivo para salvá-lo, digitar também a sua extensão. Isso vai facilitar sua localização ao usar alguns programas.
**se tiver curiosidade, abra cada um deles em um programa como Bloco de Notas para visualizar suas diferenças.
Você também pode fazer o contrário. Importar uma matriz concatenada, e exportar os diferentes fragmentos em arquivos separados. São exatamente os mesmos passos, porém selecionando diferentes formatos. O programa tem outros recursos, mas que estão além do propósito do mini-curso.
Agora que você já tem a matriz concatenada, que tal partir pra seleção de modelos ou para uma análise de máxima parcimônia? Divirta-se!