O que é a máxima parcimônia?
A máxima parcimônia é um método/filosofia que considera a árvore mais curta (em número de passos de transformação) a melhor hipótese sobre a relação filogenética de um dado conjunto de terminais. Passo de transformação é o custo atribuído à mudança de estado de caráter em um dado ramo da árvore, seja ela a mudança de uma base nucleotídica para outra, de aminoácidos, ou da cor da íris de um animal. Quanto menos transformações, mais curta será a árvore, e por isso, mais parcimoniosa. Neste método, o que os programas fazem é investigar o máximo de topologias alternativas possível (ou todas, se sua matriz tiver menos que 25 terminais; ver mais detalhes abaixo) dada a sua matriz de caracteres. Cada uma delas será mensurada, e as árvores mais curtas (ótimas) serão retidas como o resultado mais parcimonioso (veja mais em Goloboff, 1998; Wheeler, 2012).
TNT - Tree analysis using New Technology
Uma vez que você esteja com suas sequências alinhadas e concatenadas, o próximo passo é abrir o seu dataset em um software de análise filogenética. Uma das opções é o TNT: um software gratuito que lida exclusivamente com o critério da Máxima Parcimônia. Ao abrir o programa, a primeira coisa a se fazer é criar um output file (esse será um arquivo .txt que registrará todas as ações realizadas por você no programa). Sugerimos que salve todos os arquivos de uma análise em um mesmo diretório.
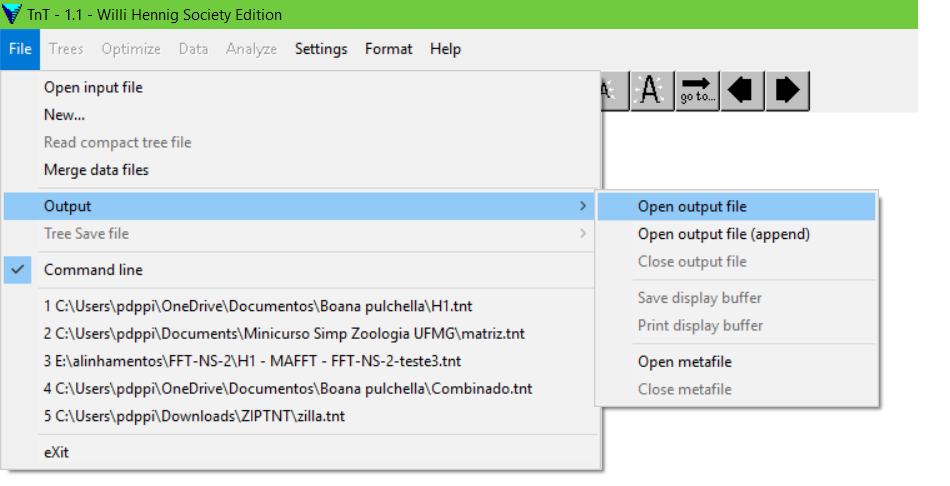
File > Output > Open Output File
Feito isso, você deverá agora, antes de abrir seu dataset, decidir como tratará os gaps das suas sequências. Provavelmente, seu alinhamento conterá gaps devido a inserções/deleções prováveis nas diferentes sequências sendo empregadas. Não é nosso objetivo neste curso discutir qual tratamento dar. Basicamente, se você considerar os gaps como “missing data”, um dado gap não contará como um passo transformacional na sua árvore, ou seja, vai contar como 0. Se você tratar os gaps como um 5º estado de transformação (sequências de DNA têm 4 estados: A, C, G, T), um dado gap vai contar como um ou mais passos na sua árvore (vai depender de você pesar ou não este estado caráter). Se você optar por tratar gaps como 5º estado, pode simplesmente abrir sua matriz, pois este é o default do TNT. Se quiser abordá-los como entrada faltante, vá em:
Format > Data Format > Read Gaps as Missing
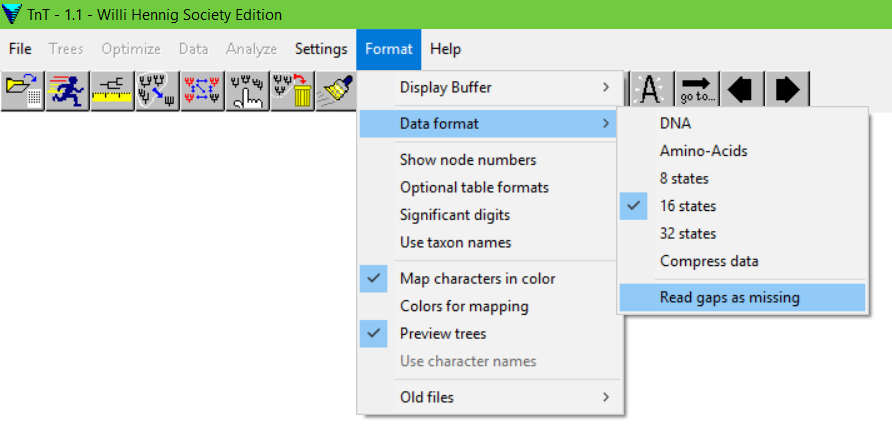
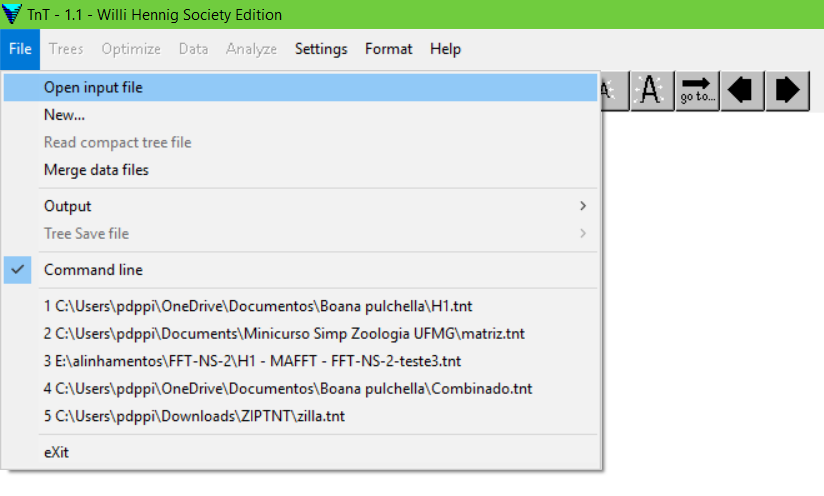
Feito isso é hora de abrir sua matriz. Pode-se fazer isso de duas maneiras:
File > Open input file
Ou ir no Display, no primeiro ícone com uma pasta aberta, com arquivos:
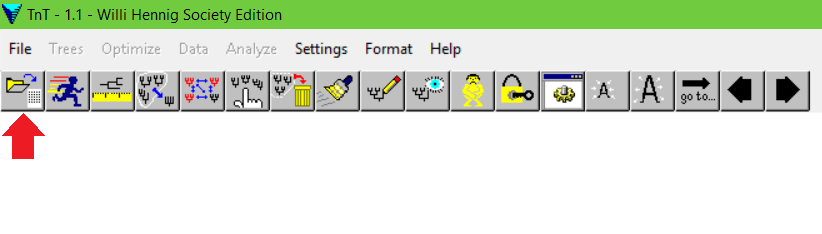
Depois de aberta a matriz, é importante definir sua raiz. Por default, o TNT reconhece o primeiro táxon da matriz como a raiz. Se você pretende usar outro, recomendamos definir antes de rodar sua análise. Vá em:
Data > Outgroup taxon
Selecione o táxon de interesse e clique em OK. A seguinte mensagem deverá aparecer na última linha do buffer:
Outgroup is taxon XXX – Espécie_qualquer
Finalmente agora vamos rodar sua análise. Existem três maneiras básicas de realizar sua busca (estamos aqui fazendo análises sem pesagem de caracteres). Se o seu conjunto de dados possui menos que 25 terminais, recomenda-se fazer uma busca por enumeração implícita (Implicit enumeration). Este método irá analisar todas as possíveis árvores existentes de acordo com o número de terminais da sua matriz e encontrar quais as topologias mais curtas. Porém, a maioria dos datasets hoje em dia lida com mais do que 25 terminais, o que torna o número de árvores possíveis completamente absurdo, e nenhum computador moderno consegue examinar todas possibilidades de árvores existentes com seu conjunto de dados. Isto é um problema NP completo (para mais a respeito Wheeler, 2012). Para esta situação pode-se fazer buscas tradicionais ou por novas tecnologias. Hoje sabemos que a segunda opção rende maiores resultados, pois foge de ilhas de resultados (para maior discussão a respeito, ver Page, 1993). Obs.: No caso de datasets extensos, recomenda-se aumentar a capacidade de memoria do programa para armazenar árvores. Assim evita-se que sua busca seja interrompida por saturação de memória. Para isso há dois caminhos: Na linha de comando (parte inferior da tela do programa) digitar:
hold XXXX
onde X é o número de árvores possíveis de se reter. Ou acessar pelo menu:
Settings > Memory
A seguinte tela aparecerá:
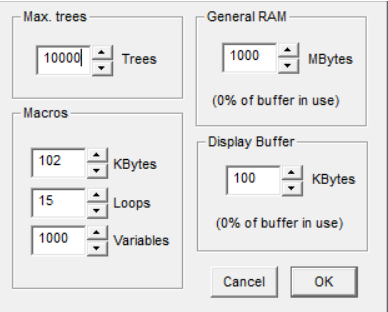
Em Max. Trees (canto superior esquerdo), digite o numero desejado (o programa tem limite de capacidade de acordo com o banco de dados a ser trabalhado.
Busca Tradicional (Traditional Search)
Analyze > Traditional search
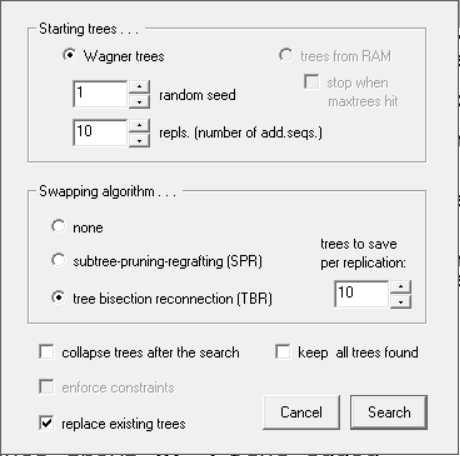
Random seed: é um número aleatório de identificação de por onde começar a busca, não significa quantidade. É como se você estivesse utilizando uma semente específica para plantar, e não uma qualquer do saco. Para você replicar sua análise, recomenda-se sempre usar o mesmo número. Importante, caso preencher com 0, o TNT será aleatório e iniciará a análise considerando o relógio do computador, e pode ser que você não chegue no mesmo resultado em futuras análises.
Repls. (number of add. Seqs.): este é o número de árvores iniciais para fazer sua busca. A partir de cada uma delas será realizada uma perturbação para encontrar outras árvores possíveis.
Swapping algorithm: é o tipo de perturbação nas árvores iniciais que o programa fará para buscar opções mais curtas ou igualmente curtas que ele encontrará na busca.
Subtree-pruning-regrafting (SPR): cada nó de uma árvore é recortado e reinserido em outras partes disponíveis e calcula-se novamente o comprimento do novo arranjo (Goloboff, 1998).
Tree bisection reconnection (TBR): similarmente ao SPR, recorta-se um nó. O pequeno grupo recortado é enraizado em todas as suas possibilidades, e as mesmas são reinseridas na árvore mãe em diferentes pontos (Goloboff, 1998).
Trees to save per replication: é o número de árvores ótimas que você deseja que o programa salve na memória por rodada de SPR ou TBR.
New Technologies Search
Diferentemente da busca tradicional, que em datasets grandes tende a ficar presa em ilhas de topologias (Nixon, 1999; Goloboff, 1999), este método de busca visa perturbar os dados afim de explorar novas ilhas de topologias (ver Page, 1993).
Analyze > New Technologies Search
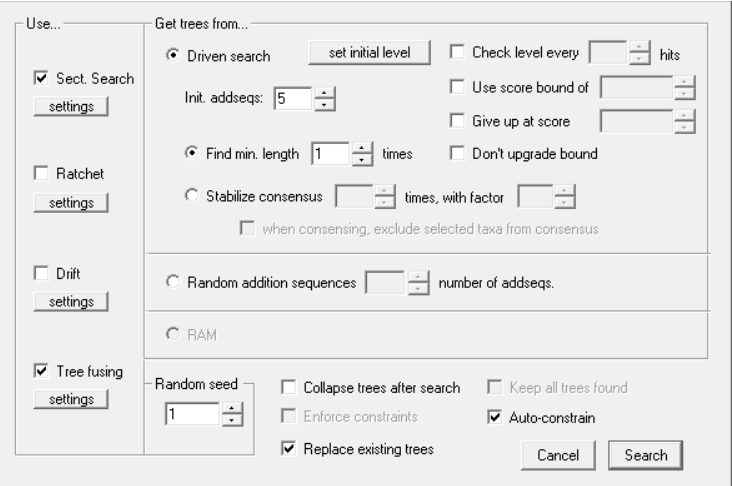
Na coluna da esquerda, existem quatro opões de ferramentas para utilizar. Você pode escolher quais deseja, nas diferentes combinações que preferir.
Sectorial Search: Basicamente pega-se um clado X, e reanalisa ele independentemente do restante da árvore. A reanálise é “recolada” na árvore original. Se o resultado obtido é mais curto que a topologia original, a nova topologia é retida (Goloboff, 1999).
Parsimony Ratchet: A partir de uma árvore inicial (árvore de Wagner), atribui-se novos pesos a caracteres de maneira aleatória (ou deleta-se caracteres aleatoriamente), realiza-se recombinação de ramos (e.g., TBR) com a matriz “repesada”, volta-se o peso dos caracteres para valores originais, e novamente outra recombinação de ramos, retendo uma ou poucas árvores. Essa sequência pode se repetir diversas vezes (Nixon, 1999).
Tree Drifting: Assim como o Ratchet, também perturba os dados. Neste caso, soluções subótimas são retidas durante as recombinações de ramos. Se a diferença relativa de fit (ver Goloboff & Farris, 2001) der próxima a um, o no rearranjo encontrado pode ser salvo (Goloboff, 1999).
Tree Fusing: Basicamente, este método pega grupos de idêntica composição taxonômica, provenientes de diferentes topologias (árvores) e os troca (Goloboff, 1999).
Randon seed: igual na busca tradicional.
Find min length: número de arvores mais curtas deseja encontrar. O programa ficará buscando usando as ferramentas que você escolheu, e enquanto busca opções mais curtas aparecerão. Então, se você deixar o valor default 1, ao fazer uma rodada de cada uma das opções de busca que marcou, ele encerrará a busca retendo as x topologias igualmente mais parcimoniosas. Quanto mais você colocar, mais rodadas das ferramentas de busca serão executadas e aumenta-se o número de topologias encontradas. Quanto mais colocar, mais robusto será seu resultado, mas mais tempo demorará sua análise.
Ao terminar uma busca, uma tela similar à seguinte deverá aparecer no seu buffer:
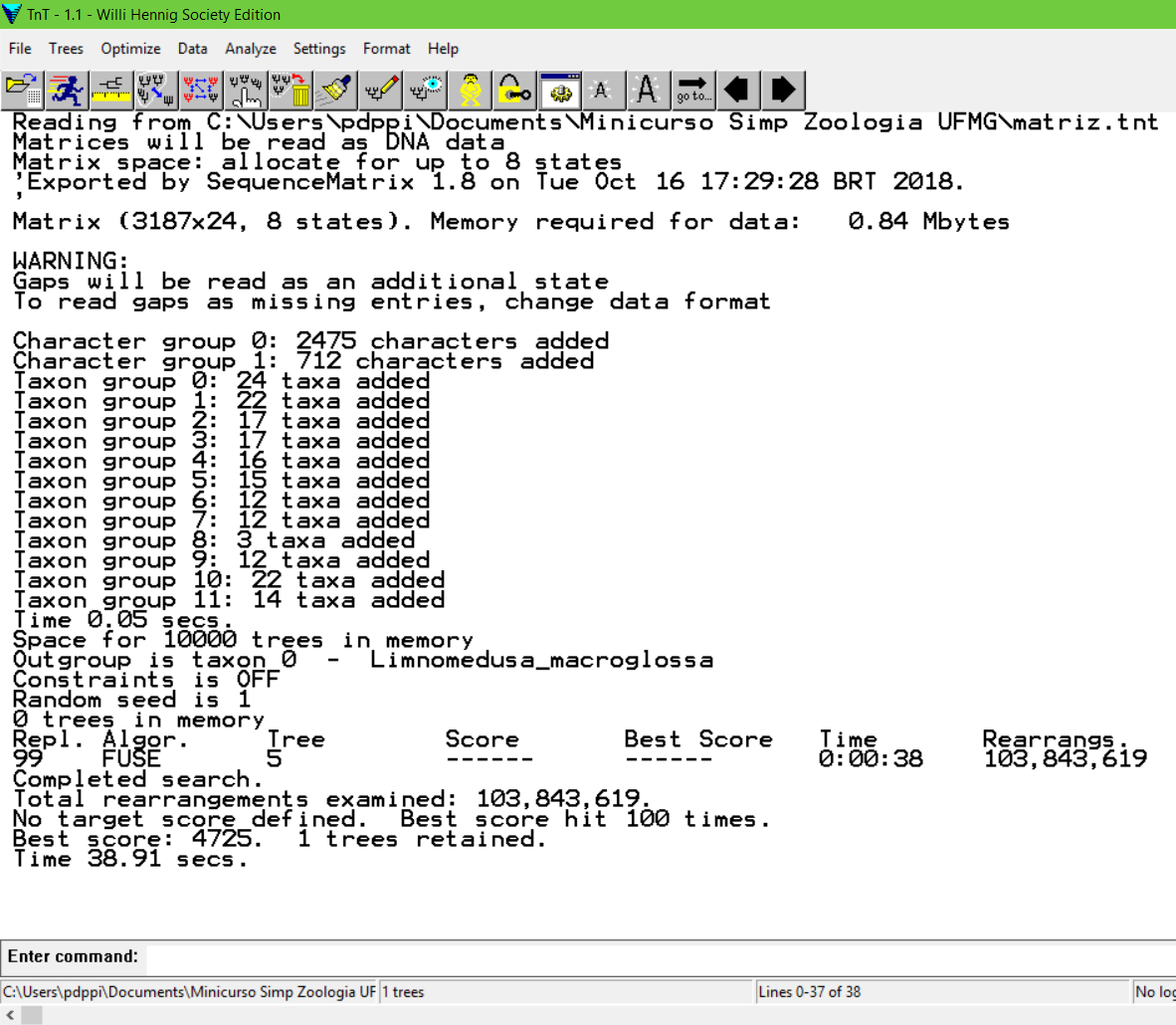
Neste caso, foi uma busca com o deffault de Traditional Search. Na penultima linha, o best score é o valor do comprimento minimo encontrado, ou seja, seu resultado mais parcimonioso. À frente dele, X trees retained, é o número de topologias encontradas que possuem tal comprimento.
Cálculo de suporte
Jackknife e Bootstrap
Ambos as medidas, são muito similares. O algorítimo irá remover caracteres aleatoriamente de sua matriz, e buscar árvores ótimas sem os mesmos, e o resultado final, ou seja, o valor de suporte de cada nó, será a porcentagem com que tal agrupamento foi reamostrada durante sua busca. A diferença entre os dois, é que o Bootstrap, além de apagar caracteres aleatoriamente, irá multiplicar outros aleatoriamente também, e isso na prática é como se você estivesse atribuindo peso a determinados caracteres. Valores de suporte abaixo de 50% são descartados ou normalmente são omitidos das topologias publicadas, por uma matemática simples: se um nó foi recuperado em 50% da reamostragem, ele também deixou de ser recuperado nos outros 50%. Assim, 50 – 50 = 0, e o suporte dele é tratado como desprezível.
Para calcular tanto o Bootstrap quanto o Jackknife, acesse:
Analyze > Resampling
A seguinte tela irá aparecer:
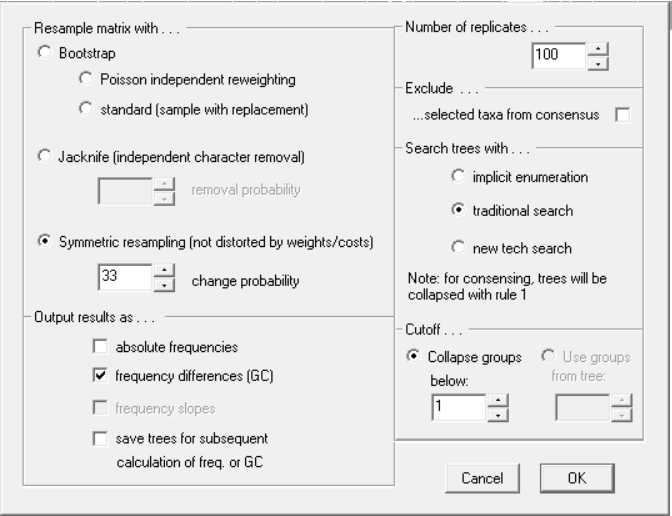
Ao escolher Bootstrap, você deverá optar por:
Poisson independent reweighting: A partir de cálculos de Poisson, estabelece vetores de peso aleatoriamente a diferentes caracteres. Ou seja, normalizando todos os caracteres ao peso zero, e o algorítimo vai incrementando pesos aos caracteres à medida que amostra os mesmos. Veja mais em Sidall, (2001).
Standard (sample with replacement): A matriz será reamostrada de maneira que alguns caracteres serão removidos, e outros serão multiplicados, para que o tamanho final da matriz seja igual ao inicial. Veja mais em Wheeler (2012).
No Jackknife irá informar a probabilidade de remoção, que nada mais é a probabilidade que cada caráter da matriz tem de ser removido na reamostragem.
Para ambos os métodos, você deverá informar qual tipo de resultado espera ser apresentado:
Absolute frequencies: Valor absoluto da frequência em que um dado clado foi reamostrado no cálculo. Em geral se considera apenas valores acima de 50% (0.5), pois um se o grupo foi reamostrado em apenas 50% das árvores, nos outros 50% ele não existe. Logo, 0.5- 0.5 = 0. Isso leva a alguns questionamentos sobre o uso de frequências absolutas como medidas de suporte. Veja mais em Goloboff et al. (2003).
Frequency diferences (GC): É outro tipo de cálculo, que leva em consideração a frequência com que o grupo é recuperado (G) e as vezes em que é contradito (C). Porém, pode levar a falsas ideias de suporte em casos onde topologias mais parcimoniosas são contraditórias (isto é, o grupo colapsa no consenso), porém uma parte dos caracteres sustenta um grupo ou outro. É mais utilizado com reamostragem simétrica, que está fora do escopo deste curso. Veja mais em Goloboff et al. (2003). Ou ambos.
Para um suporte razoável, é comum fazer cálculos de 1000 réplicas (canto superior direito), e optar por um método de busca, que será similar à busca pela topologia mais curta (para alterar os parâmetros de busca, você deve clicar sobre “traditional search” ou “new tech search”, e um menu similar ao de busca por topologias mais parcimoniosas irá surgir). Obviamente, para minimizar o tempo, com New Technology Search, por exemplo, uma busca encontrando 10 vezes o valor mais curto, dependendo do seu banco de dados, já será bem válida. Raciocínio similar se aplica a busca tradicional.
Ao terminar o cálculo, imagem similar à seguinte irá aparecer na tela do seu computador:
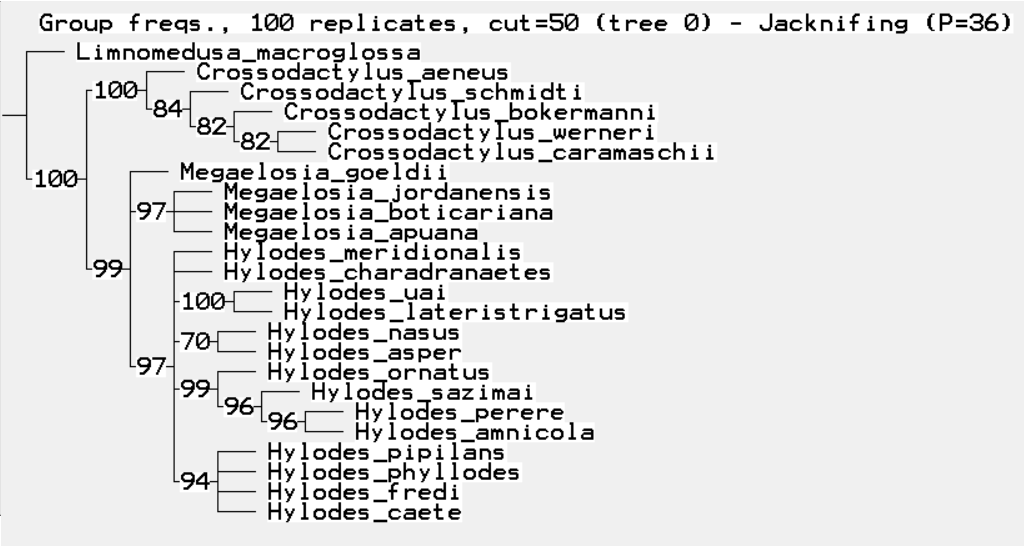
Antes de qualquer coisa, o primeiro botão que você deve pressionar é o “M” do seu teclado. Assim abrirá uma caixa de diálogo para você salvar um arquivo metafile com o resultado da sua reamostragem de suporte. Se você apertar “ESC”, por exemplo, essa tela sairá, e você perderá os seus resultados. E acredite: você não quer isso! Um calculo de suporte em geral é bem demorado!
Para calcular quaisquer destes dois índices, você necessita apenas da sua matriz. Não é necessário ter as árvores mais curtas em mãos. Mas, a topologia resultante dos índices de reamostragem, não necessariamente corresponderá às suas árvores mais curtas. Ela apenas te dá os suportes dos nós mais comuns.
Para você obter os valores, através do TNT, em uma de suas árvores mais curtas, você deverá primeiro fazer a busca, ou ter as árvores já salvas em formato .ctf. No primeiro caso, ao calcular as arvores, não as apague da memória RAM. Inicie o procedimento normal de reamostragem dito acima, e na tela dos índices, no canto inferior direito, selecione a opção “use groups from tree”, e selecione uma de suas árvores na barra de rolagem que será habilitada. Assim, a árvore com valores de suporte que aparecerá ao final, será uma das suas mais parcimoniosas, ou seu consenso por exemplo. Aperte “M” e salve o metafile!
Se você já tiver as árvores salvas em .ctf, abra sua matriz no TNT, depois vá em:
File > read compact tree file
e abra o seu arquivo. Vá em:
Analyze > resampling
e também ative o “use groups from tree” conforme descrito no parágrafo acima.
Suporte de Bremer
O suporte de Bremer é um índice que avalia quantos passos mais comprida a sua árvore deveria ser, para que um dado nó colapse. Sendo assim, quanto maior o valor de suporte, significa que são necessárias mais transformações para derrubar tal clado (veja mais em Goodman et al., 1982; Bremer, 1988). Para ver isso, o método irá analisar topologias subótimas, isto é, topologias X passos mais compridas do que o resultado ótimo encontrado em suas buscas. Isso significa ampliar consideravelmente o número de topologias avaliadas para calcular o suporte do clado. Então, pensando em matrizes muito grandes, já é possível imaginar que o tempo computacional de calcular o suporte de Bremer de todas as topologias ótimas do seu resultado, pode ser muito custoso. Por isso, usa-se antes de mais nada calcular o consenso estrito dos seus resultados de busca, e então fazer o cálculo de Bremer somente sobre ele. Aqui são válidas duas observações: 1) o consenso estrito somente deverá ser calculado no caso de mais de uma árvore igualmente parcimoniosa encontrada; 2) agrupamentos ausentes no consenso estrito terão valor de Bremer = 0.
Para calcular o consenso, existem duas maneiras, pelo menu superior, ou pelo menu interativo. Pelo menu superior:
Trees > Consensus
E então a seguinte tela irá aparecer:
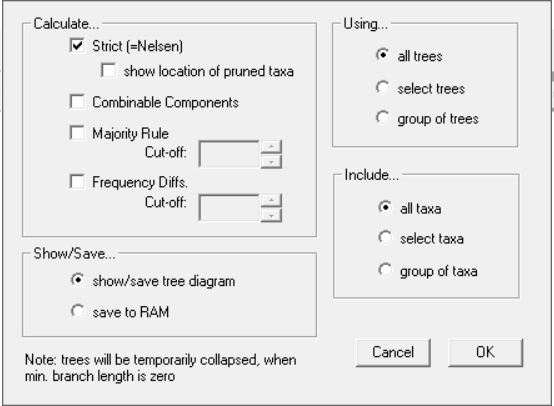
Nela, mantenha a caixa “Strict” marcada, em “Calculate…”, em “Using…” e “Include…” mantenha também o default, isto é, “all trees” e “all taxa” selecionados (a não ser que você queira avaliar algo especificamente). Para ver seu consenso, em “Show/Save…” selecione “show/save tree diagram”. Clique em OK, e uma árvore aparecerá na tela do seu computador. Para salvá-la em metafile, assim como no Jackknife e Bootstrap, aperte “M”, e você poderá salvar a figura do seu consenso.
Porém, para dar seguimento ao cálculo de Bremer, você deverá selecionar a opção “save to RAM” em “Show/Save…”. Isto garantirá que tal arranjo ficará armazenado na memória do programa.
A outra opção para calcular o consenso é o Menu Interativo. Basta ir na opção indicada na figura abaixo, e seguir os mesmos passos descritos acima.
Um,a vez calculado o consenso, vá em:
Trees > Tree Buffer > Select trees
Ou no Menu interativo em:
A seguinte janela irá aparecer:
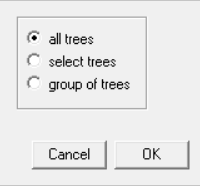
Clique em select trees, e a seguinte janela irá aparecer:
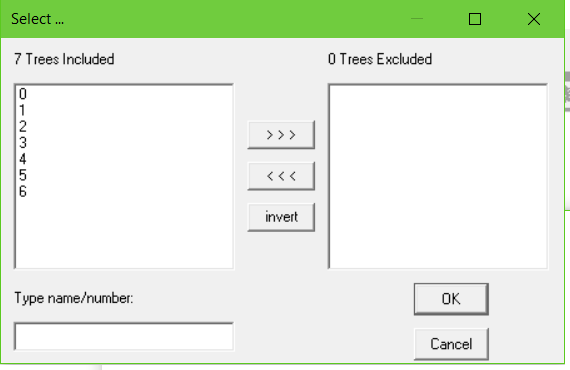
Quando você salvou seu consenso estrito, deverá ter notado que surgiu a seguinte mensagem no Buffer do programa: “Strict consensus of Z trees (0 taxa excluded) calculated, as tree X.” Este X, é o número de identificação da última árvore dos seus resultados. Sendo assim, na janela acima, você deverá selecionar doas as árvores da lista da esquerda, exceto a última (em “X trees included”) e clicar no botão »>. E depois clique em OK. E outra vez em OK na janela remanescente. Dessa maneira a única árvore que ficou incluída deverá ser o seu consenso calculado.
Agora, voltemos ao menu:
Trees>Bremer Supports
A seguinte tela irá aparecer:
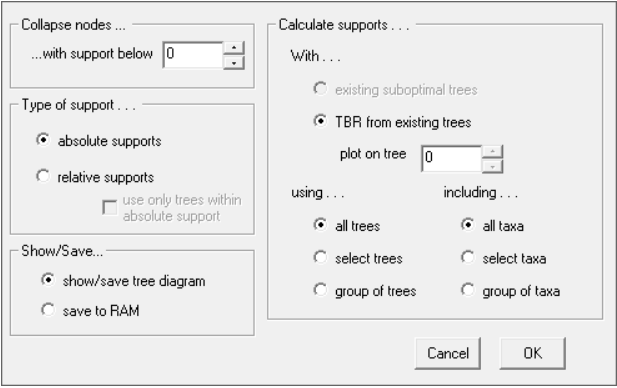
Mantenha tudo no default. Como você já selecionou anteriormente o consenso, verá que no menu da direita “Calculate supports…” a opção “TBR from existing trees plot on tree 0” é a única possível. Sua árvore X se transformou em árvore 0 por que você excluiu as demais para a análise. E, apesar de não ser tão indutivo, você agora DEVE clicar sobre “TBR from existing trees plot on tree 0”. O seguinte menu irá aparecer:
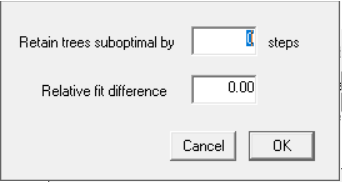
Como não estamos trabalhando aqui com pesagem implícita, ignore o campo “Relative fit difference” e mantenha-o com o valor 0. No campo superior, “Retain trees suboptimal by” você deverá colocar o número de passos a mais que pretende testar, para achar os valores de Bremer dos seus nós. E aqui se começa um processo de tentativa e erro. Se iniciar por exemplo com um valor de, por exemplo, 15, e clicar em OK nos dois menus, seu resultado aparecerá como um diagrama na tela (para salvá-lo em Metafile, novamente aperte “M”). Você provavelmente verá que, com 15 passos a mais, muitos ramos terão um valor anotado. Este é o valor de Bremer do nó acima dele. Porém, em outros ramos, o valor 15? (acrescido de uma interrogação) estará presente. Isso significa que ao buscar árvores 15 passos mais compridas do que as suas, você ainda não foi capaz de colapsar todos os nós. Sendo assim, você deverá calcular o Bremer novamente, e colocar um valor maior do que 15 em “Retain trees suboptimal by”. E esse processo se repetirá até você atingir valores sem interrogação em todos os ramos.
Agora que vimos o básico de uma análise de máxima parcimônia, vamos aprender um pouco sobre como preparar uma análise de máxima verossimilhança, e em seguida uma inferência Bayesiana!
Referências
Bremer, K. 1988. The limits of amino acid sequence data in Angiosperm phylogenetic reconstruction. Evolution, 42(4): 795–803.
Goloboff, P. A. 1998. Princípios Básicos de Cladística. Sociedad Argentina de Botánica. 81.
Goloboff, P. A. 1999. Analyzing large data sets in reasonable times: solutions for composite optima. Cladistics, 15: 415–428.
Goloboff, P. A.; Farris, J. S. 2001. Methods for quick consensus estimation. Cladistics, 17: S26–S34.
Goloboff, P. A.; Farris, J. S.; Källersjö, M.; Oxelman, B.; Ramírez, M. J.; Szumik, C. A. 2003. Improvements to resampling measures of group support. Cladistics, 19: 324–332.
Goodman, M; Olson, C.; Beeher, J.; Czelusniak, J. 1982. New perspectives in the molecular biological analysis of mammalian phylogeny. Acta Zoologica Fennica, 169: 19–35.
Nixon, K. C. 1999. The Parsimony Ratchet, a new method for rapid parsimony analysis. Cladistics, 15: 407–414.
Page, R. D. M. 1993. On islands of trees and the efficacy of different methods of branch swapping in finding most-parsimonious trees. Systematic Biology, 42(2): 200–210.
Siddall, M. E. 2001. Measures of Support. Em: Methods and Tools in Biosciences and Medicine. DeSalle, R.; W. C. Wheeler & G. Giribet (eds.). Basel: Birkhäuser Verlag. Cap. 5, pp. 80–101.
Wheeler, W. C. 2012. Systematics: a course of lectures. Malaysia: Wiley-Backwell. 426.