RAxML - Randomized Axelerated Maximum Likelihood
Existem vários softwares para inferir árvores filogenéticas utilizando o critério da máxima verossimilhança, como por exemplo o garli (Zwickl, 2006), o PhyML (Guindon et al. 2010) e o TREE-PUZZLE (Schmidt et al. 2002). No entanto, para o nosso curso abordaremos o programa RaxML (Stamatakis, 2014), por ser um software rápido, simples de usar e bastante utilizado. Já existe inclusive uma versão do RaxML para dados de sequenciamento de alto rendimento, chamado RAxML Next Generation.
Uso local
O input do RAxML é feito com arquivos fasta ou phylip, sendo o segundo a melhor opção (já tive problemas tentando fazê-lo ler o primeiro). Então vamos aos passos:
1) Pegue seu arquivo hylodidae.phy e coloque-o na pasta em que vai analisá-lo. 2) O RAxML suporta inferir os parâmetros de maneira diferente para as diferentes partições. Lembra do nosso resultado do PartitionFinder2? Pois é, foram 4 as partições definidas. Vá até o seu editor de texto favorito (bloco de notas, notepad, etc.) e escreva o seguinte:
DNA, h1 = 1-2475
DNA, coi_pos1 = 2476-3187\3
DNA, coi_pos2 = 2477-3187\3
DNA, coi_pos3 = 2478-3187\3
Salve este arquivo como partitions.txt e coloque-o na mesma pasta em que está hylodidae.phy.
3) Agora abra o prompt de comando (ou terminal, dependendo do seu OS) e vá até o diretório em que estão seus arquivos phylip e das partições. No windows, geralmente ao abrir o prompt de comando ele estará no diretório C:\Users\nome_de_usuario. Então, supondo que seus arquivos estejam na pasta raxml dentro da pasta Documents, digite:
cd Documents\raxml
4) Uma vez no diretório com os arquivos, pegue a versão do RAxML do seu computador (no nosso exemplo é raxmlHPC-PTHREADS-SSE3) e arraste para o prompt de comando. Aperte a tecla espaço e escreva os comandos (antes de teclar enter leia a explicação dos comandos abaixo):
-f d -p 12345 -m GTRGAMMA -N 2 -q partitions.txt -o Limnomedusa_macroglossa -s hylodidae.phy -n ml -T 2
-f d - Usa o algoritmo new rapid hill-climbing (Stamatakis et al., 2007) para buscar a árvore mais verossímil.
-p 12345 - A busca pela árvore mais verossímil se inicia com uma árvore de parcimônia, e para calcular esta árvore você precisa definir uma random seed. O número depois de -p poderia ter sido qualquer um. Este parâmetro é obrigatório em qualquer análise do RAxML que necessite algum grau de aleatorização
-m GTRGAMMA - Este comando especifica o modelo de substituição nucleotídica, no nosso caso, GTR + G. O RAxML não suporta diferentes modelos em diferentes partições. Segundo Stamatakis (vide manual), ele decidiu assim pois os outros modelos são casos específicos do modelo General Time-Reversible (GTR; Tavaré, 1986) e, como o RAxML é rápido o suficiente, não era necessário implementar os modelos mais simples (e menos custosos computacionalmente). Este parâmetro é obrigatório em qualquer análise, se não estiver presente o RAxML apresentará um erro.
-N 2 - Este parâmetro define que se busque a árvore mais verossímil duas vezes. Quanto maior este número, melhor será o resultado final mas mais lenta será a busca.
-q partitions.txt - Este parâmetro mostra ao programa qual é o arquivo em que estão definidas nossas partições.
-o Limnomedusa_macroglossa - Este parâmetro mostra ao RAxML onde vamos enraizar a árvore. Ao contrário das análises de parcimônia, não é necessário especificar este parâmetro.
-s hylodidae.phy - Este parâmetro especifica sua matriz. Também é um parâmetro obrigatório!
-n ml - este parâmetro especifica o nome dos seus arquivos de saída, que no nosso caso virão com a extensão .ml.
-T 2 - Este parâmetro é muito importante, porque na versão PTHREADS especifica o número de processadores que vamos utilizar. Se colocarmos mais do que temos, a análise tende a ser mais lenta. Se colocarmos mais do que precisamos, também. Para a nossa análise, 2 é o número ótimo, mesmo que você tenha mais processadores disponíveis.
5) Agora que conseguimos a árvore mais verossímil, hora de calcular o suporte da nossa árvore. Faremos isso utilizando réplicas de bootstrap não-paramétricas, de forma parecida a que usamos no programa TNT. Arraste sua versão do programa para o terminal e digite:
-b 12345 -p 12345 -m GTRGAMMA -N 20 -q partitions.txt -o Limnomedusa_macroglossa -s hylodidae.phy -n bootstrap -T 2
-b 12345 - Este parâmetro liga o bootstrap e especifica um *random seed** pra ele. Note que de acordo com o parâmetro -N faremos 20 réplicas de bootstrap, o que é muito pouco. Porém, fazer 1000 réplicas pode demorar bastante.
6) Agora é hora de ver com que frequência os clados da sua árvore mais verossímil aparecem nas suas réplicas de bootstrap. Arraste novamente sua versão do programa para o prompt, aperte espaço e digite:
-f b -m GTRGAMMA -t RAxML_bestTree.ml -z RAxML_bootstrap.bootstrap -o Limenomedusa_macroglossa -s nome_do_arquivo.phy -n tre -T 2
-t RAxML_bestTree.ml - Este parâmetro indica que o arquivo RAxML_bestTree.ml contem a árvore mais verossímil.
-z RAxML_bootstrap.bootstrap - Este parâmetro indica que o arquivo RAxML_bootstrap.bootstrap contem suas réplicas de bootstrap.
Note que, mesmo sem utilizarmos a matriz original ou um modelo de substituição nucleotídica, é obrigatório que se inclua estes parâmetros através dos comandos -m e -s, ou o programa não funciona.
Pronto! Agora você possui a árvore com as réplicas de bootstrap e pode abri-la através do programa FigTree. O arquivo se chama RAxML_bipartitions.tre.
Uso em servidor de alta performance (plataforma CIPRES)
O CIPRES Science Gateway é uma plataforma pública que disponibiliza recursos computacionais para a inferência de árvores filogenéticas. É bastante poderosa e é limitada a algumas milhares de horas de processamento por ano. A primeira coisa que você deve fazer para usar o CIPRES Science Gateway é criar uma conta. Depois disso é só aproveitar sua estrutura computacional e os softwares instalados! Vamos utilizar a versão do RAxML disponível no portal.
1) Após logar na sua conta, clique em Create New Folder e então dê um nome (obrigatório) e uma descrição (opcional) para seu diretório.
2) Agora dentro do seu novo diretório, vá em Data e depois clique em Upload/Enter Data. Então, clique no botão Escolher arquivos, faça o upload de hylodidae.phy e partitions.txt.
3) Com os arquivos necessários já enviados para o CIPRES, entre em Tasks e então clique em Create New Task. A seguinte tela irá aparecer:
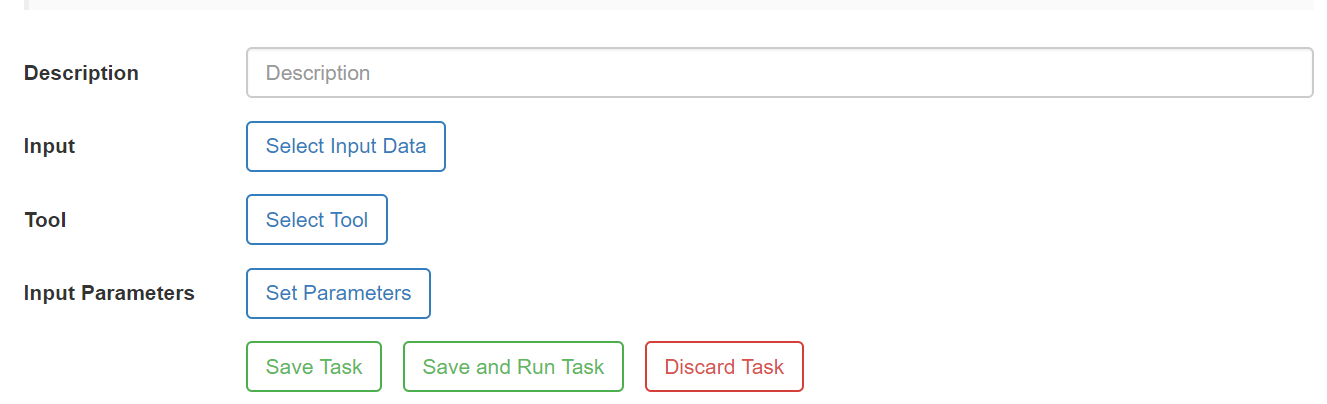
4) Vá em Select Input Data, selecione nossa matriz e clique em Select Data.
5) Clique em Select Tool e selecione a opção RAxML-HPC v.8 on XSEDE. Depois, clique onde está escrito 36 Parameters Set.
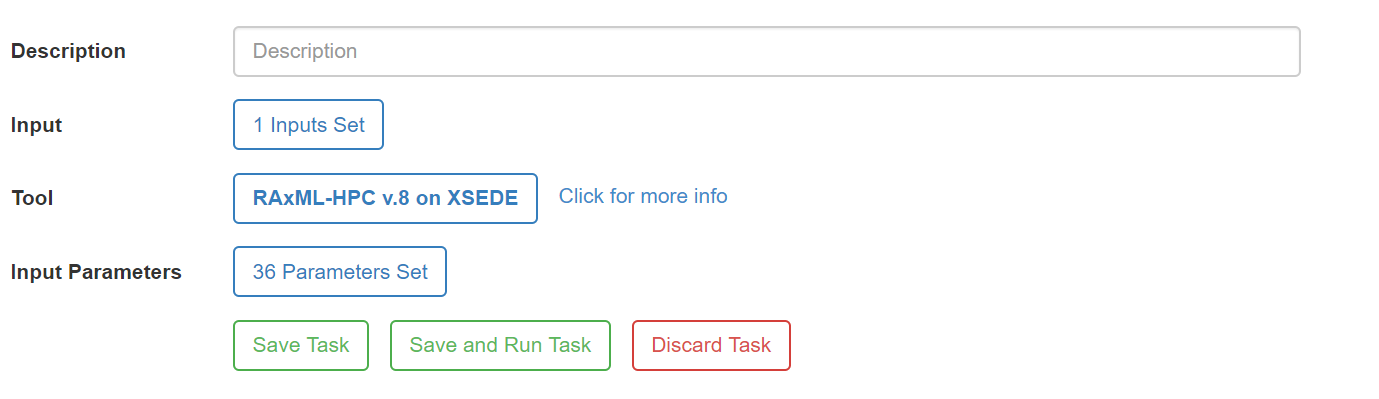
6) Vamos escolher os parâmetros da nossa busca. O RAxML tem uma opção de busca de árvore mais verossímil seguida de um rápido bootstrap, muito boa para grandes matrizes ou para resultados rápidos (nosso caso). Primeiro, vamos colocar tre em Set a name for output file, mudar o Outgroup (-o) para Limnomedusa_macroglossa e mudar o parâmetro Use a mixed/partitioned model? (-q) para conter nosso arquivo partitions.txt. Então clique em Advanced Parameters.
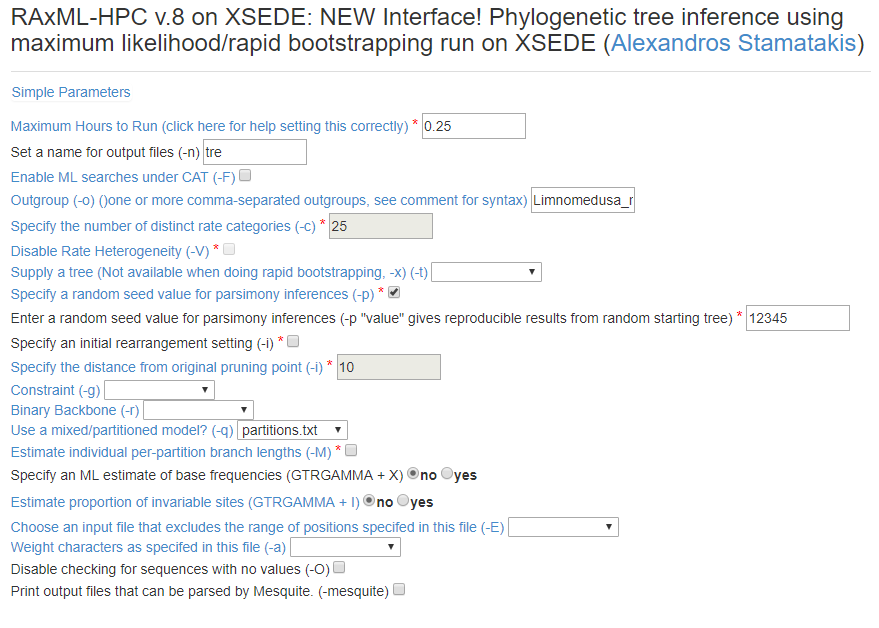
Agora, em Nucleic Acid Options selecione GTRGAMMA:
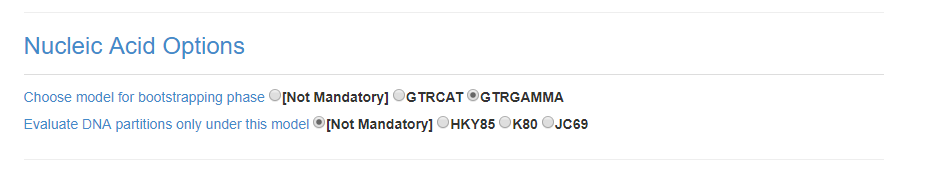
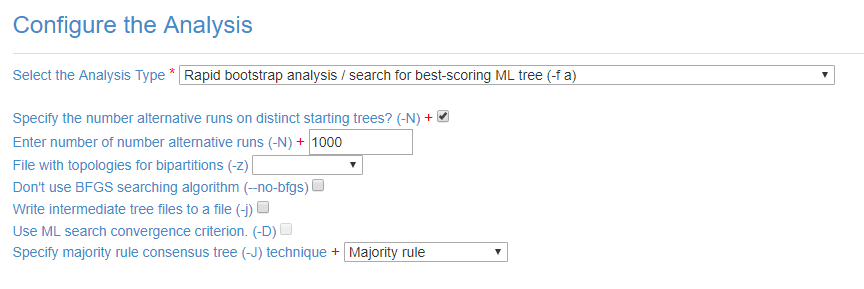
Em Configure the Analysis, selecione a opção -f a, marque a caixinha com a opção -N e mude para 1000 o número de réplicas:
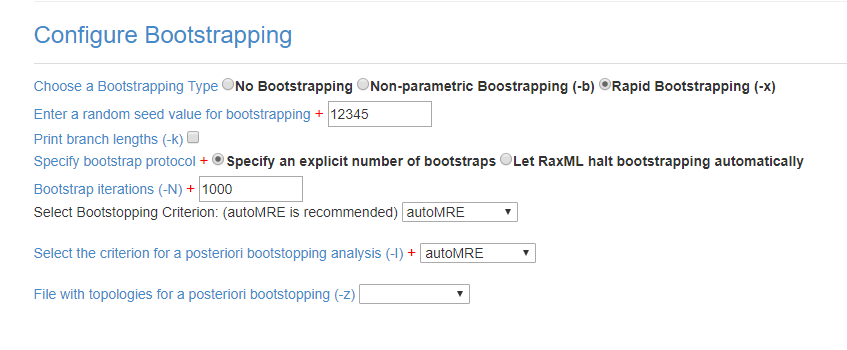
Em Configure Bootstraping, escolha a opção Rapid Bootstraping (-x), marque a opção Specify an explicit number of bootstraps e coloque 1000 Bootstrap iterations (-N):
Agora clique em Save Parameters e depois em Save and Run Task. Agora é só aguardar que você receberá um email quando a análise estiver pronta!
Vamos ao último critério de optimalidade que vamos falar no curso: inferência Bayesiana!
Referências
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. (2010). New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology, 59:307-321.
Schmidt HA, Strimmer K, Vingron M, von Haeseler A. (2002). TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics, 18:502-504.
Stamatakis A, Blagojevic F, Nikolopoulos DS, Antonopulos, CD. (2007). Exploring New Search Algorithms and Hardware for Phylogenetics:
RAxML Meets the IBM Cell. The Journal of VLSI Signal Processing Systems for Signal, Image, and Video Technology 48:271-286.
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30:1312–1313.
Tavare S. (1986). Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences (American Mathematical Society), 17:57–86.
Zwickl DJ. (2006). Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation, The University of Texas at Austin.