Seleção de modelos e particionamento da matriz
Para os métodos filogenéticos de máxima verossimilhança e inferência Bayesiana, antes de começarmos nossas análises, é necessário escolher o modelo de substituição nucleotídica que mais se encaixa nos nossos dados. Estes modelos descrevem as diferentes probabilidades de mudança de um nucleotídeo para outro ao longo de uma árvore filogenética. O uso de um modelo ou outro pode afetar os resultados de uma análise filogenética, por isso devemos escolher o melhor modelo com muito cuidado! Além disso, modelos mais simples, apesar de serem computacionalmente menos custosos (deixando a análise mais rápida) podem fazer com que um clado pareça mais bem suportado do que ele na verdade é. Quanto mais complexo (i.e., mais parâmetros ele tem) é um modelo molecular, mais computacionalmente custoso ele é. Então, a escolha do modelo molecular é importante porque deve ser uma razão entre custo computacional e adequabilidade do modelo aos dados.
Outra coisa importante é que o genoma não evolui de maneira homogênea, então é interessante que possamos adequar os modelos moleculares a cada uma das partições que formos analisar (normalmente cada um dos genes da nossa análise). Para estimar o modelo de evolução mais adequado aos nossos dados, usaremos os programas jModelTest2 e PartitionFinder2.
jModelTest2
O jModelTest2 tem uma versão em linha de comando, normalmente utilizada em clusters e supercompudadores com sistemas operacionais sem interface gráfica e uma versão com interface gráfica (ou GUI, graphical user interface). Como vamos utilizar o programa em nossos computadores pessoais, utilizaremos a versão GUI. Primeiramente, vá até o diretório em que descomprimiu o jModelTest2 e abra o arquivo jModelTest.jar. A pártir de agora as instruções e imagens serão baseadas no manual do programa, disponível aqui.
Vamos começar pelo nosso fragmento COI. Carregue o arquivo hylodidae_coi_alinhado.fas:
File > Load DNA alignment
Depois, vá até:
Analysis > Compute Likelihood Scores
E então selecione os modelos a serem computados. Para que gastemos um tempo razoável (poucos minutos), vamos restringir o número de esquemas de substituição para 11. Marque os outros parâmetros como na figura abaixo e depois é só apertar o botão Compute Likelihoods. Lembrando que o número de processadores varia conforme o modelo do computador.
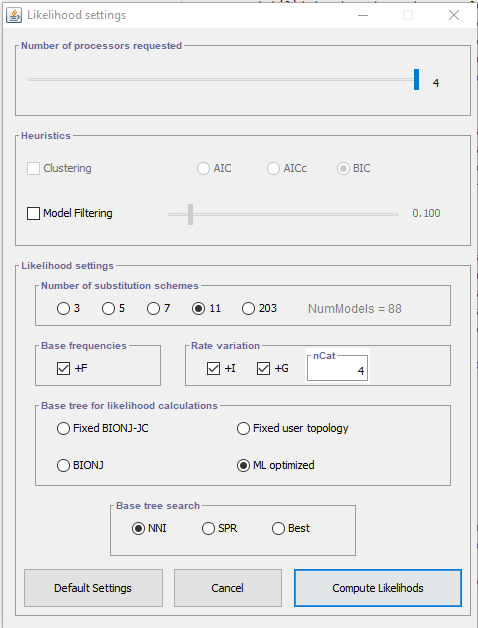
Agora que computamos as verossimilhanças, vamos selecionar os modelos estatisticamente. Existem alguns critérios para isto, mas no nosso curso utilizaremos o Bayesian Information Criterion (BIC; Schwarz, 1978). Então vá em:
Analysis > Do BIC calculations
Uma janela se abrirá, e então aperte o botão Do BIC calculations. Os resultados serão mostrados no console. Para ver melhor os resultados, vá em:
Results > Show results table
O melhor modelo será aquele com menor valor de BIC, no nosso caso, TrN + G + I. Se você quiser, pode salvar os resultados no formato HTML e pode abri-lo com seu navegador. Para isso, vá em:
Results > Build HTML log
Repita o processo com as sequências de 16S. Você espera que o resultado seja o mesmo?
PartitionFinder2
O PartitionFinder2 é um programa muito interessante, porque além de fazer a seleção do melhor modelo molecular, ele também divide suas partições da melhor maneira possível. Quanto mais dividida sua matriz estiver, mais parâmetros os programas de filogenia terão de calcular e maior será o tempo computacional para realizar a tarefa. Então, você divide suas partições no maior número de fragmentos possível e o PF2 analisa seus dados e vê o que pode ser analisado na mesma partição, poupando tempo computacional. Agora veremos como usar esse programa.
1) A primeira coisa que devemos fazer é preparar a nossa matriz, transformando-a em um arquivo phylip. Acabamos de fazer isso na parte de concatenamento, lembram? Você precisará do seu bom arquivo hylodidae.phy.
2) O PF2 não possui interface gráfica. Então, todos os comandos que queremos que o programa realize deverão ser escritos em um arquivo que o programa vai ler enquanto estiver sendo executado. Este arquivo deve obrigatoriamente se chamar partition_finder.cfg e existe uma versão modelo dele dentro do diretório onde você instalou o PF2, na pasta:
examples/nucleotide
Abra o arquivo em um editor de texto (bloco de notas, notepad ou similar). Vamos fazer algumas alterações para que fique como o arquivo abaixo:
## ALIGNMENT FILE ##
alignment = hylodidae.phy;
## BRANCHLENGTHS: linked | unlinked ##
branchlengths = linked;
## MODELS OF EVOLUTION: all | allx | mrbayes | beast | gamma | gammai | <list> ##
models = mrbayes;
# MODEL SELECCTION: AIC | AICc | BIC #
model_selection = aicc;
## DATA BLOCKS: see manual for how to define ##
[data_blocks]
h1 = 1-2475;
coi_pos1 = 2476-3187\3;
coi_pos2 = 2477-3187\3;
coi_pos3 = 2478-3187\3;
## SCHEMES, search: all | user | greedy | rcluster | rclusterf | kmeans ##
[schemes]
search = all;
Como vocês podem ver, mudamos o nome do arquivo de entrada, vamos estimar apenas os modelos que existem no programa mrbayes, dividimos nosso dataset em 4 partições (explico a seguir*) e vamos procurar todos os esquemas de partições possíveis, o que num dataset como o nosso é factível. Existem algoritmos de busca heurística que o PF2 usa para matrizes com maior número de partições, mas não é nosso caso. Evite usar ´search = all´ se sua matriz possui mais de 10 partições ou você pode não obter o resultado nesta encarnação. Quanto mais partições, maior a combinação de esquemas possível.
*Como o COI é um gene codificante e uma mudança em cada uma das três posições do códon pode gerar aminoácidos completamente diferentes, podemos supor que cada uma dessas posições evolui de forma independente. Quando escrevemos nos data_blocks ‘2476-3187\3’, estamos dizendo: “Parta da posição 2476 e vá até a posição 3187, considerando um nucleotídeo a cada 3 bases”.
3) Crie uma pasta e coloque os arquivos hylodidae.phy e partitionfinder.cfg nessa pasta. Agora abra o prompt de comando do windows (ou o terminal se estiver utilizando um OS Unix) e escreva ‘python’ e aperte espaço. Agora vá até o diretório onde você instalou o PF2, pegue o arquivo PartitionFinder.py e arraste para o prompt de comando (ou terminal) e aperte novamente espaço. Agora pegue a pasta que você criou com os dois arquivos e arraste para o prompt, e então aperte enter. No seu console vai aparecer uma tela mais ou menos assim:
INFO | 2018-12-12 20:01:49,765 | ------------- PartitionFinder 2.1.1 -----------------
INFO | 2018-12-12 20:01:49,796 | You have Python version 2.7
INFO | 2018-12-12 20:01:49,836 | Command-line arguments used: C:\Users\Pedro Taucce\Documents\partitionfinder-2.1.1\PartitionFinder.py C:\Users\Pedro Taucce\Dropbox\curso_ufmg\modelo
INFO | 2018-12-12 20:01:49,861 | ------------- Configuring Parameters -------------
INFO | 2018-12-12 20:01:49,861 | Setting datatype to 'DNA'
INFO | 2018-12-12 20:01:49,861 | Setting phylogeny program to 'phyml'
INFO | 2018-12-12 20:01:49,861 | Program path is here C:\Users\Pedro Taucce\Documents\partitionfinder-2.1.1\programs
INFO | 2018-12-12 20:01:49,861 | Setting working folder to: 'C:\Users\Pedro Taucce\Dropbox\curso_ufmg\modelo'
INFO | 2018-12-12 20:01:49,891 | Loading configuration at '.\partition_finder.cfg'
INFO | 2018-12-12 20:01:49,950 | Setting 'alignment' to 'hylodidae.phy'
INFO | 2018-12-12 20:01:50,020 | Setting 'branchlengths' to 'linked'
INFO | 2018-12-12 20:01:50,065 | You set 'models' to: mrbayes
INFO | 2018-12-12 20:01:50,128 | This analysis will use the following 24 models of molecular evolution
INFO | 2018-12-12 20:01:50,128 | JC, K80, SYM, F81, HKY, GTR, JC+G, K80+G, SYM+G, F81+G, HKY+G, GTR+G, JC+I, K80+I, SYM+I, F81+I, HKY+I, GTR+I, JC+I+G, K80+I+G, SYM+I+G, F81+I+G, HKY+I+G, GTR+I+G
INFO | 2018-12-12 20:01:50,128 | Setting 'model_selection' to 'aicc'
INFO | 2018-12-12 20:01:50,144 | Setting 'search' to 'all'
INFO | 2018-12-12 20:01:50,144 | ------------------------ BEGINNING NEW RUN -------------------------------
INFO | 2018-12-12 20:01:50,144 | Looking for alignment file '.\hylodidae.phy'...
INFO | 2018-12-12 20:01:50,158 | Using 4 cpus
INFO | 2018-12-12 20:01:50,158 | Beginning Analysis
INFO | 2018-12-12 20:01:50,292 | Reading alignment file '.\hylodidae.phy'
INFO | 2018-12-12 20:01:50,306 | Reading alignment file '.\analysis\start_tree\source.phy'
INFO | 2018-12-12 20:01:50,354 | Starting tree will be estimated from the data.
INFO | 2018-12-12 20:01:50,354 | Estimating Maximum Likelihood tree with RAxML fast experimental tree search for .\analysis\start_tree\filtered_source.phy
INFO | 2018-12-12 20:01:50,385 | Using a separate GTR+G model for each data block
Agora é só esperar uns 10 a 15 minutos e ver a mágica acontecer.
4) Agora que a análise terminou, vá até a pasta que você criou e veja o arquivo best_scheme.txt e veja quais são suas partições e quais os modelos escolhidos! Ou então baixe o arquivo best_scheme.txt
Pronto! Agora que já tem os modelos de substituição nucleotídica, pode fazer uma análise de máxima verossimilhança ou de inferência Bayesiana. Mas primeiro vamos fazer uma análise de máxima parcimônia?
Referências
Schwarz GE. (1978). Estimating the dimension of a model. Annals of Statistics, 6: 461–464