Aquisição de sequências no GenBank
O que é o GenBank?
O GenBank é uma base de dados que contem todas as sequências de DNA disponíveis publicamente. Quando você elabora um trabalho utilizando sequências de DNA, provavelmente você irá visitar o GenBank para adquirir sequências antes de começar suas análises e muito provavelmente irá acessá-lo depois para depositar as sequências que produziu: é obrigatório depositar as sequências num repositório público para ter um artigo publicado. Uma funcionalidade muito útil do GenBank é o BLAST, onde você pode pegar uma sequência e comparar com TODAS as sequências disponíveis na imensa base de dados do GenBank. É muito útil, por exemplo, para saber se a sequência em questão está contaminada.
Usando o BLAST
Como dito anteriormente, a ferramenta BLAST compara a sequência desejada com todas as sequências disponíveis no banco de dados do GenBank. Para utilizá-lo, primeiro baixe esta sequência de onça pintada. Depois, abra-a no bloco de notas, notepad ou programa equivalente. Agora vá até o site do BLAST e então, copie (ctrl + c) a sequência baixada, e logo depois cole na janela correspondente e então aperte o botão “BLAST”, como na figura abaixo:
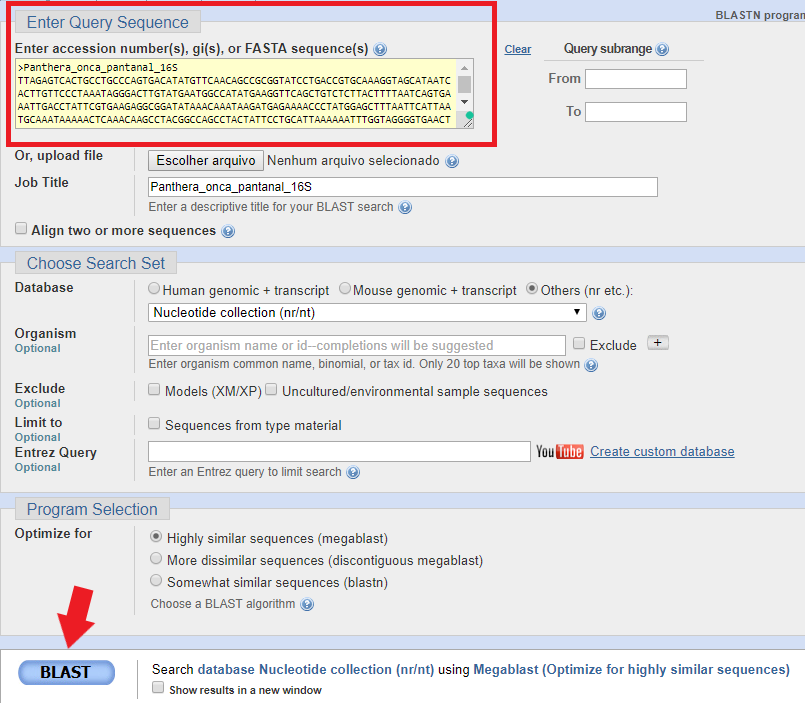
Outra maneira de usar o BLAST é colocando como input o arquivo todo, clicando no botão “Escolher arquivo.
Qual foi a sequência mais próxima à nossa? Se você tivesse acabado de produzir esta sequência, o que isto poderia significar?
Usando a plataforma R para adquirir sequências
Se todas as sequências de DNA públicas já produzidas estão no GenBank, é lá que vamos quando queremos adquirir alguma sequência. Experimente navegar no site: busque pela sequência de algum organismo e de um fragmento. Se estiver sem ideia, vamos ver sequências de rã-de-unhas-africana (Xenopus laevis) para o fragmento 16S. Digite no campo de busca:
Xenopus laevis 16S
E então aperte o botão search. A página irá te mostrar todas as sequências disponíveis de Xenopus laevis que contenham o marcador mitocondrial 16S rRNA. No nosso caso mais de 600 sequências. Baixar tudo isso na base do clique daria um trabalho danado! Por isso alguns pesquisadores fizeram pacotes da plataforma R que interagem com o GenBank.
Então vamos supor que você encontrou uma espécie nova de sapo que parece ser do gênero Hylodes e queira fazer uma árvore incluindo dois marcadores mitocondriais, 16S rRNA e COI, da família de anfíbios Hylodidae para posicionar sua nova espécie. Essa família inclui mais dois gêneros além de Hylodes: Megaelosia e Crossodactylus. Daria bastante trabalho baixar todas as sequências disponíveis no GenBank para a família no clique, então vamos fazer utilizando a plataforma R!
Preparando o R
Primeiramente é necessário baixar os pacotes, que no nosso caso serão APE (Paradis et al., 2004) e rentrez (winter, 2017). Para isso, vá até o console do R e digite:
install.packages("ape")
install.packages("rentrez")
Depois cheque se os pacotes foram instalados corretamente, digitando:
"ape" %in% rownames(installed.packages())
"rentrez" %in% rownames(installed.packages())
Se a resposta para ambos os comandos for “TRUE”, então você pode ir adiante.
Baixando as sequências por nome
Continuando com sua ideia da filogenia. Então você quer todas as sequências de Hylodes, Megaelosia e Crossodactylus dos marcadores mitocondriais 16S e COI. Então, no console do R, digite o seguinte (lembrando de trocar o caminho dentro do comando setwd para uma pasta existente):
library("ape")
library("rentrez")
setwd("C:/caminho/para/pasta/desejada")
for (i in c("Hylodes", "Megaelosia", "Crossodactylus")){
terms <- paste(i, " [Organism]"," AND 16S [ALL]")
search <- entrez_search(db="nuccore", term=terms, retmax=10000)
if (search$count > 0){
seqs <- entrez_fetch(db="nuccore", id=search$ids, rettype="fasta")
write(seqs, file = paste(i, "hylodidae_16S.fasta", sep="_"), sep="\n", append = TRUE)
}
}
for (i in c("Hylodes", "Megaelosia", "Crossodactylus")){
terms <- paste(i, " [Organism]"," AND coi [ALL]")
search <- entrez_search(db="nuccore", term=terms, retmax=10000)
if (search$count > 0){
seqs <- entrez_fetch(db="nuccore", id=search$ids, rettype="fasta")
write(seqs, file = paste(i, "hylodidae_coi.fasta", sep="_"), sep="\n", append = TRUE)
}
}
Com isso espera-se que, na pasta desejada, você tenha dois arquivos: hylodidae_16S.fasta e hylodidae_coi.fasta. Cada um deles vai conter todas as sequências de Hylodidae para o marcador escolhido. Agora é só editar os nomes e se divertir!
Baixando as sequências por GenBank id
Suponhamos que acaba de sair uma nova espécie de Megaelosia, e os autores do trabalho fizeram uma ótima árvore, a qual você gostaria de usar e apenas adicionar sua espécie nova de Hylodes para posicioná-la. Infelizmente os autores não disponibilizaram a matriz utilizada no artigo, mas no material suplementar existem todas as GenBank ids utilizadas para construir a matriz. Nesse caso você pode baixar do GenBank todas as sequências deles, utilizando o seguinte código:
library("ape")
library("rentrez")
setwd("C:/caminho/para/pasta/desejada")
ids.16s=c("KY007117.1", "KU215902.1", "KM390791.1", "AY843579.1", "AY263235.1", "MF624225.1",
"KY627916.1", "KJ961575.1", "KJ961577.1", "MF624238.1", "AY843689.1", "KJ961586.1",
"KJ961578.1", "DQ283072.1", "KJ961583.1", "KM390793.1", "KM390794.1", "KU495250.1",
"KY002953.1", "KY627911.1", "KY627913.1", "KY627914.1")
seqs <- entrez_fetch(db="nuccore", id=ids.16s, rettype="fasta")
write(seqs, file = "16S_ids.fasta", sep="\n", append = FALSE)
ids.coi=c("KU494387.1", "DQ502738.1", "KU494457.1", "KC593345.1", "KJ961562.1", "KJ961561.1",
"KJ961558.1", "KJ961557.1", "KJ961556.1", "DQ502873.1", "KJ961565.1", "KU494594.1",
"KJ961566.1", "DQ502839.1")
seqs <- entrez_fetch(db="nuccore", id=ids.coi, rettype="fasta")
write(seqs, file = "coi_ids.fasta", sep="\n", append = FALSE)
Agora você tem no seu computador os arquivos 16S_ids.fasta e coi_ids.fasta com as sequências desejadas. Agora que você possui as sequências, hora de partir para o alinhamento!