Seleção de modelos e particionamento da matriz
Para os métodos filogenéticos de máxima verossimilhança e inferência Bayesiana, antes de começarmos nossas análises, é necessário escolher o modelo de substituição nucleotídica que mais se encaixa nos nossos dados. Estes modelos descrevem as diferentes probabilidades de mudança de um nucleotídeo para outro ao longo de uma árvore filogenética. O uso de um modelo ou outro pode afetar os resultados de uma análise filogenética, por isso devemos escolher o melhor modelo com muito cuidado! Além disso, modelos mais simples, apesar de serem computacionalmente menos custosos (deixando a análise mais rápida) podem fazer com que um clado pareça mais bem suportado do que ele na verdade é. Quanto mais complexo (i.e., mais parâmetros ele tem) é um modelo molecular, mais computacionalmente custoso ele é. Então, a escolha do modelo molecular é importante porque deve ser uma razão entre custo computacional e adequabilidade do modelo aos dados.
Outra coisa importante é que o genoma não evolui de maneira homogênea, então é interessante que possamos adequar os modelos moleculares a cada uma das partições que formos analisar (normalmente cada um dos genes da nossa análise). Para estimar o modelo de evolução mais adequado aos nossos dados, usaremos o programa PartitionFinder2.
PartitionFinder2
O PartitionFinder2 é um programa muito interessante, porque além de fazer a seleção do melhor modelo molecular, ele também divide suas partições da melhor maneira possível. Quanto mais dividida sua matriz estiver, mais parâmetros os programas de filogenia terão de estimar e maior será o tempo computacional para realizar a tarefa. Então, você divide suas partições no maior número de fragmentos possível e o PF2 analisa seus dados e vê o que pode ser analisado na mesma partição, poupando tempo computacional. Agora veremos como usar esse programa.
1) A primeira coisa que devemos fazer é o download da nossa matriz. Já preparei uma matriz da família de anfíbios anuros Hylodidae contendo partes dos genes mitocondriais COI, 12S rRNA, tRNAval e 16S rRNA, que você pode baixar aqui. O PF2, assim como o RAxML, utiliza arquivos phylip (.phy).
2) Abra o arquivo hylodidae.phy em um editor de texto e dê uma olhadela. O que você vê? Quantos terminais são? Quantos pares de base?
3) O PF2 não possui interface gráfica. Então, todos os comandos que queremos que o programa realize deverão ser escritos em um arquivo que o programa vai ler enquanto estiver sendo executado. Este arquivo deve obrigatoriamente se chamar partition_finder.cfg. Se você tem o programa instalado em seu computador, existe uma versão modelo dele dentro do diretório onde você instalou o PF2, na pasta:
examples/nucleotide
Caso não possua o programa instalado, abra um editor de texto (bloco de notas, notepad ou similar) e escreva o conteúdo abaixo, salvando o arquivo como partition_finder.cfg.
## ALIGNMENT FILE ##
alignment = infile.phy;
## BRANCHLENGTHS: linked | unlinked ##
branchlengths = linked;
## MODELS OF EVOLUTION: all | allx | mrbayes | beast | gamma | gammai | <list> ##
models = mrbayes;
# MODEL SELECCTION: AIC | AICc | BIC #
model_selection = aicc;
## DATA BLOCKS: see manual for how to define ##
[data_blocks]
h1 = 1-2475;
coi_pos1 = 2476-3187\3;
coi_pos2 = 2477-3187\3;
coi_pos3 = 2478-3187\3;
## SCHEMES, search: all | user | greedy | rcluster | rclusterf | kmeans ##
[schemes]
search = all;
Lembrando que, se você for fazer a análise em outro computador que não o do CIPRES, onde está escrito “alignment =” deve estar escrito exatamente o nome da sua matriz ( no nosso caso, seria hylodidae.phy). Está escrito “infile.phy” pois é uma exigência do portal CIPRES.
Como vocês podem ver, mudamos o nome do arquivo de entrada, vamos estimar apenas os modelos que existem no programa mrbayes, dividimos nosso dataset em 4 partições (explico a seguir*) e vamos procurar todos os esquemas de partições possíveis, o que num dataset como o nosso é factível. Existem algoritmos de busca heurística que o PF2 usa para matrizes com maior número de partições, mas não é nosso caso. Evite usar ´search = all´ se sua matriz possui mais de 10 partições ou você pode não obter o resultado nesta encarnação. Quanto mais partições, maior a combinação de esquemas possível.
*Como o COI é um gene codificante e uma mudança em cada uma das três posições do códon pode gerar aminoácidos completamente diferentes, podemos supor que cada uma dessas posições evolui de forma independente. Quando escrevemos nos data_blocks ‘2476-3187\3’, estamos dizendo: “Parta da posição 2476 e vá até a posição 3187, considerando um nucleotídeo a cada 3 bases”.
Uso do PartitionFinder2 em servidor de alta performance (plataforma CIPRES)
O CIPRES Science Gateway é uma plataforma pública que disponibiliza recursos computacionais para a inferência de árvores filogenéticas. É bastante poderosa e é limitada (caso queiram usar gratuitamente) a 1000 de horas de processamento por ano para usuários que não estão nos EUA. A primeira coisa que você deve fazer para usar o CIPRES Science Gateway é criar uma conta. Depois disso é só aproveitar sua estrutura computacional e os softwares instalados! Vamos utilizar a versão do PartitionFinder2 disponível no portal.
1) Após logar na sua conta, clique em Create New Folder e então dê um nome (obrigatório) e uma descrição (opcional) para seu diretório.
2) Agora dentro do seu novo diretório, vá em Data e depois clique em Upload/Enter Data. Então, clique no botão Escolher arquivos, faça o upload de hylodidae.phy e partition_finder.cfg.
3) Com os arquivos necessários já enviados para o CIPRES, entre em Tasks e então clique em Create New Task. A seguinte tela irá aparecer:
4) Vá em Select Input Data, selecione nossa matriz e clique em Select Data.
5) Clique em Select Tool e selecione a opção PartitionFinder2 on XSEDE. Depois, clique onde está escrito 10 Parameters Set.
6) Vamos escolher os parâmetros da nossa corrida. Por ser uma análise relativamente pequena, o tempo default de meia hora será mais que suficiente para terminá-la. Não se esqueça de selecionar o arquivo partition_finder.cfg na parte Select cfg file (you can also create one below). Se você clicar em Advanced Parameters pode também criar o arquivo de configuração.
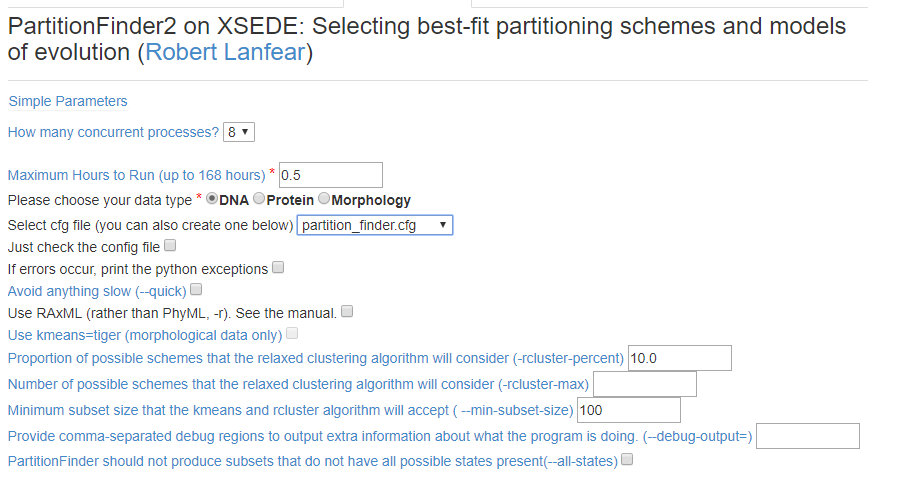
Agora clique em Save Parameters e depois em Save and Run Task. Agora é só aguardar que você receberá um email quando a análise estiver pronta!
Pronto! Agora que já tem os modelos de substituição nucleotídica, podemos fazer a análise de máxima verossimilhança.
RAxML - Randomized Axelerated Maximum Likelihood
Existem vários softwares para inferir árvores filogenéticas utilizando o critério da máxima verossimilhança, como por exemplo o garli (Zwickl, 2006), o PhyML (Guindon et al. 2010) e o TREE-PUZZLE (Schmidt et al. 2002). No entanto, para o nosso curso abordaremos o programa RaxML (Stamatakis, 2014), por ser um software rápido, simples de usar e bastante utilizado. Já existe inclusive uma versão do RaxML para dados de sequenciamento de alto rendimento, chamado RAxML Next Generation.
Uso em servidor de alta performance (plataforma CIPRES)
1) Após logar na sua conta, clique em Create New Folder e então dê um nome (obrigatório) e uma descrição (opcional) para seu diretório.
2) O RAxML suporta inferir os parâmetros de maneira diferente para as diferentes partições. Lembra do nosso resultado do PartitionFinder2? Pois é, foram 4 as partições definidas. Vá até o seu editor de texto favorito (bloco de notas, notepad, etc.) e escreva o seguinte:
DNA, h1 = 1-2475
DNA, coi_pos1 = 2476-3187\3
DNA, coi_pos2 = 2477-3187\3
DNA, coi_pos3 = 2478-3187\3
Salve este arquivo como partitions.txt
3) Agora dentro do seu novo diretório, vá em Data e depois clique em Upload/Enter Data. Então, clique no botão Escolher arquivos, faça o upload de partitions.txt.
4) Com os arquivos necessários já enviados para o CIPRES, entre em Tasks e então clique em Create New Task. A seguinte tela irá aparecer:
4) Vá em Select Input Data, selecione nossa matriz e clique em Select Data.
5) Clique em Select Tool e selecione a opção RAxML-HPC v.8 on XSEDE. Depois, clique onde está escrito 36 Parameters Set.
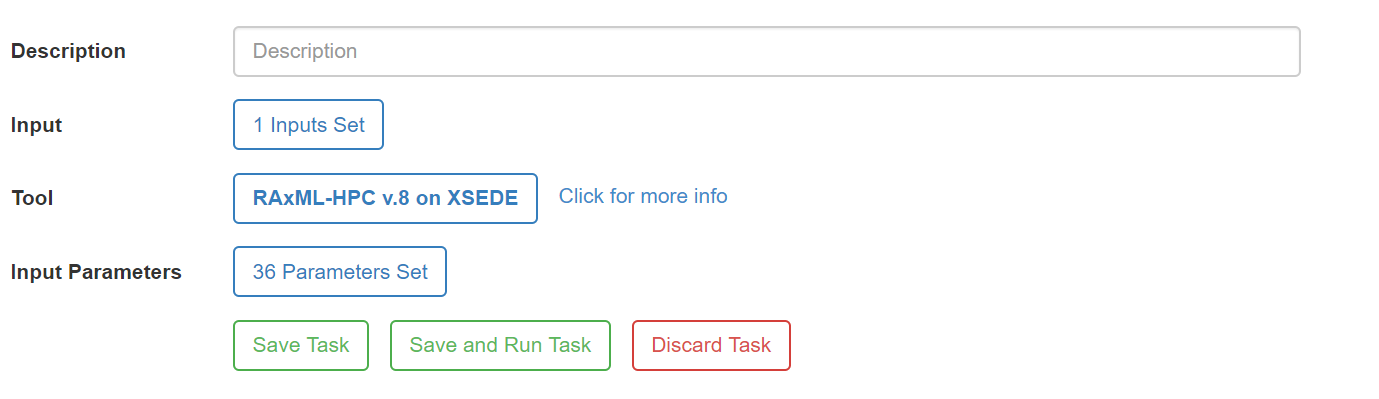
6) Vamos escolher os parâmetros da nossa busca. O RAxML tem uma opção de busca de árvore mais verossímil seguida de um rápido bootstrap, muito boa para grandes matrizes ou para resultados rápidos (nosso caso). Primeiro, vamos colocar tre em Set a name for output file, mudar o Outgroup (-o) para Limnomedusa_macroglossa e mudar o parâmetro Use a mixed/partitioned model? (-q) para conter nosso arquivo partitions.txt. Então clique em Advanced Parameters.
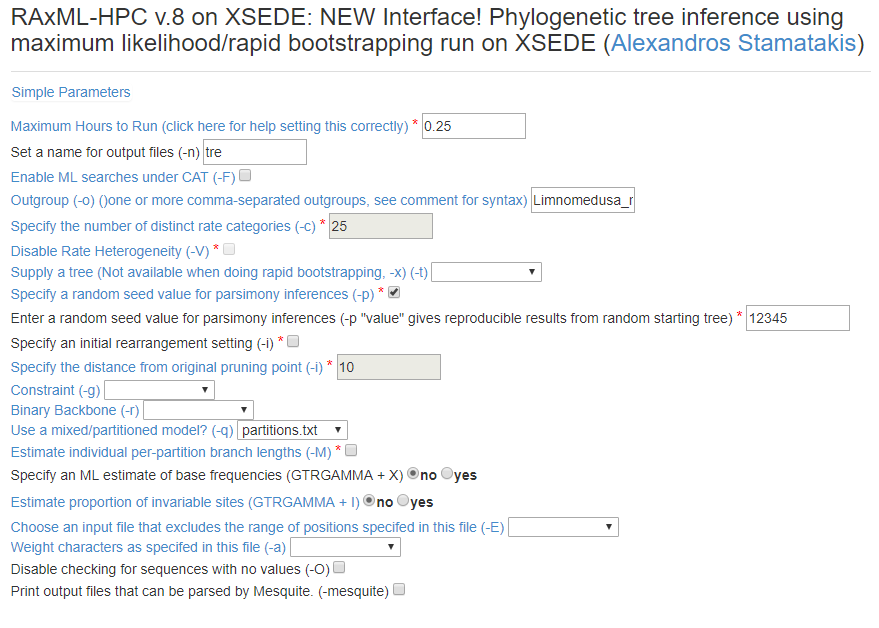
Agora, em Nucleic Acid Options selecione GTRGAMMA:
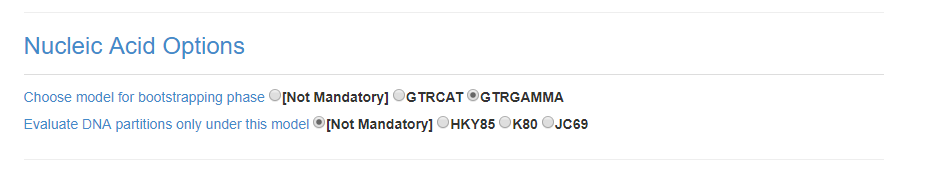
Em Configure the Analysis, selecione a opção -f a, marque a caixinha com a opção -N e mude para 1000 o número de réplicas:
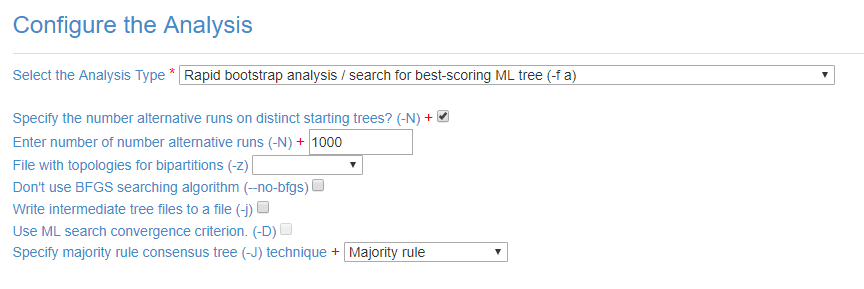
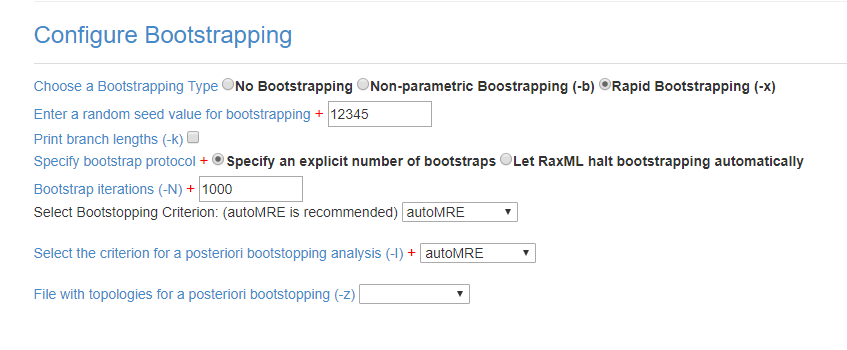
Em Configure Bootstraping, escolha a opção Rapid Bootstraping (-x), marque a opção Specify an explicit number of bootstraps e coloque 1000 Bootstrap iterations (-N):
Agora clique em Save Parameters e depois em Save and Run Task. Agora é só aguardar que você receberá um email quando a análise estiver pronta!
Referências
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. (2010). New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology, 59:307-321.
Schmidt HA, Strimmer K, Vingron M, von Haeseler A. (2002). TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics, 18:502-504.
Schwarz GE. (1978). Estimating the dimension of a model. Annals of Statistics, 6: 461–464
Stamatakis A, Blagojevic F, Nikolopoulos DS, Antonopulos, CD. (2007). Exploring New Search Algorithms and Hardware for Phylogenetics:
RAxML Meets the IBM Cell. The Journal of VLSI Signal Processing Systems for Signal, Image, and Video Technology 48:271-286.
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30:1312–1313.
Tavare S. (1986). Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences (American Mathematical Society), 17:57–86.
Zwickl DJ. (2006). Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation, The University of Texas at Austin.